
Abstract
Training machine learning models for tasks such as de novo sequencing or spectral clustering requires large collections of confidently identified spectra. Here we describe a dataset of 2.8 million high-confidence peptide-spectrum matches derived from nine different species. The dataset is based on a previously described benchmark but has been re-processed to ensure consistent data quality and enforce separation of training and test peptides.
Similar content being viewed by others


Background & Summary
De novo sequencing of proteomics tandem mass spectrometry data, in which observed fragmentation spectra are translated into corresponding peptide sequences, has been an open challenge for more than 40 years1. Recently, as in many other areas of science, considerable progress toward solving this challenge has been made using deep learning, in which multi-layer neural networks with millions of parameters are trained to generate peptide sequences from observed spectra. The first such deep learning method, DeepNovo2, has been followed by at least 22 additional publications (reviewed in3).
The standard method for evaluating these de novo sequencing methods is to use a gold standard produced via database search. In this approach, mass spectrometry data derived from a single species is searched against the reference proteome for that species, yielding a ranked list of peptide-spectrum matches (PSMs). Including in the peptide database a collection of reversed or shuffled “decoy” peptides provides a rigorous way to set a threshold in this list of PSMs while controlling the false discovery rate (FDR) among the PSMs above the threshold4. The resulting set of high-confidence PSMs can be used either to train or evaluate a de novo sequencing model.
Some version of the above protocol has been used to develop labeled training and validation data for essentially every published deep learning de novo sequencing method. One exception is methods that use spectra from synthesized peptide sequences for training5,6,7. However, even in these cases, a gold standard derived from database search is used for evaluation of the method.
Unfortunately, creating a high quality gold standard set of labeled spectra can be tricky. One challenge is ensuring that the search strategy employs appropriate parameters. For instance, one widely used benchmark dataset2 used a search strategy that failed to account for missassigned isotopic peaks during the acquisition stage. This error led to frequently assigning a deamidation modification, when the observed mass shift was better explained by an isotopic mass shift on the precursor m/z3. A second challenge relates to the notion of train/test leakage, in which information used to train the model leaks into the evaluation procedure. In the de novo setting, a common mistake is to randomly segregate a given set of labeled spectra into training and test sets, without regard to the associated peptides. As a result, spectra generated by the same peptide sequence may occur in both the training and test sets. Such duplicated peptides give an unfair advantage to the sequencing method, and the leakage will be even more useful to parameter-rich methods that are capable of memorizing many features of the training data.
In this work, we revisit the nine-species benchmark dataset that was employed in the first deep learning de novo sequencing method, DeepNovo2. This is a widely used dataset, which has been employed for training or evaluation in at least 15 subsequent studies6,8,9,10,11,12,13,14,15,16,17,18,19,20,21. The setup is quite straightforward. The authors downloaded nine publicly available datasets, all of which were generated on a Thermo Scientific Q Exactive mass spectrometer, and each of which was carried out in a different species. Each dataset was searched against the corresponding reference proteome, using a target-decoy strategy to accept a set of PSMs subject to a PSM-level FDR threshold of 1%. Because the data are derived from different species, the peptides in each set are largely (but not entirely) disjoint. To use the benchmark, it is typical to apply a cross-validation strategy, in which a model is trained on eight species and tested on the held-out species, and the procedure is repeated nine different ways.
In developing our Casanovo de novo sequencing model, we identified several problems with the nine-species benchmark21. These included the deamidation problem mentioned above, as well as some uncertainty regarding how the FDR was controlled. Perhaps most importantly, we recognized that a non-negligible proportion of peptides are shared among the different species, with the highest overlap between human and mouse.
In light of these difficulties, we downloaded the same datasets from the PRIDE repository and systematically reanalyzed all of the data, using a standard search procedure—the Tide search engine22 followed by Percolator23 with PSM-level FDR control at 1%. We then filtered the PSMs to prevent any peptide sequence from appearing in more than one species. The resulting data set was used to evaluate Casanovo21. Finally, because some of the single-species datasets are markedly larger than others, we produced a more balanced version of the dataset. Hence, we make publicly available both versions of this dataset: the peptide-disjoint dataset that can be used to avoid train/test leakage (“main”), and the reduced peptide-disjoint dataset if you want your analysis to run more quickly (“balanced”). In addition, we make available all of the intermediate files, for use in validating the benchmark.
Methods
Data sets
For our benchmark, we used the same nine studies originally identified by Tran et al.2.
-
1.
Paiva et al. investigated the protein expression response of the cowpea plant (Vigna unguiculata) to infection by Cowpea severe mosaic virus (CPSMV) by carrying out label-free proteomic analysis of cowpea leaves that were inoculated with CPSMV compared to mock inoculation controls24.
-
2.
Nevo et al. studied a rare autosomal recessive lysosomal storage disorder, cystinosis, by carrying out SILAC proteomic analysis of engineered mouse cell lines that harbor a known pathogenic mutation of the causative gene, CTNS25.
-
3.
Cassidy et al. evaluated two different analytical approaches for carrying out full proteome analysis while identifying short open reading frames: a high/low pH reversed phase LC-MS bottom-up approach and a semi-top-down strategy involving separation of proteins in a GelFree system followed by digestion and LC-MS analysis26. The experiments were carried out using the methane producing archaeon Methanosarcina mazei.
-
4.
Reuss et al. carried out proteomic analyses on a series of minimized strains of the model bacterium, Bacillus subtilis, with genomes reduced by ~ 36%27.
-
5.
Petersen et al. performed proteomic analysis of Candidatus endoloripes, which are bacterial symbionts of the Lucinidae family of marine bivalves28.
-
6.
Mata et al. characterized the proteome of the tomato pericarp at its ripe red stage29.
-
7.
Seidel et al. analyzed the global proteomic stress response in wildtype and two yeast knockout strains for the gene PBP130.
-
8.
Hu et al. studied honeybees that exhibit a suite of behaviors (Varroa sensitive hygiene—VSH) associated with infection with the Varroa destructor virus31. Proteomic analysis was carried out on mushroom bodies and antennae of adult honeybees with and without VSH.
-
9.
Cypryck et al. characterized extracellular vesicles released from human primary macrophages after infection with influenza A viruses32.
All nine studies were performed using a Thermo Scientific Q Exactive mass spectrometer.
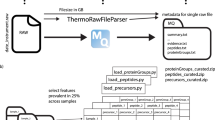
We downloaded the RAW files from the corresponding PRIDE projects (Table 1) and converted them to MGF format using the ThermoRawFileParser v1.3.433. We downloaded the corresponding nine UniProt reference proteomes and constructed a Tide index for each one, using Crux version 4.2. Note that, for one species (Vigna mungo) no reference proteome is available, so we used the proteome of the closely related species Vigna radiata.
Database search and FDR control
We assigned peptide labels to spectra using the Tide search engine followed by post-processing with Percolator. In creating the Tide index, we specified Cys carbamidomethylation as a static modification and allowed for the following variable modifications: Met oxidation, Asn deamidation, Gln deamidation, N-term acetylation, N-term carbamylation, N-term NH3 loss, and the combination of N-term carbamylation and NH3 loss by using the tide-index options --mods-spec 1M+15.994915, 1N+0.984016, 1Q+0.984016 --nterm-peptide-mods-spec 1X+42.010565, 1X+43.005814, 1X-17.026549, 1X+25.980265 --max-mods 3. Note that one of the nine experiments (Mus musculus) was performed using SILAC labeling, but we searched without SILAC modifications and hence include in the benchmark only PSMs from unlabeled peptides. Tide automatically added to each index a shuffled decoy peptide corresponding to each target peptide. Thereafter, each MGF file was searched against the corresponding index using the precursor window size and fragment bin tolerance specified in the original study (Table 1). The search engine employed XCorr scoring with Tailor calibration34, and we allowed for 1 isotope error in the selection of candidate peptides. All search results were then analyzed jointly per species using the Crux implementation of Percolator, with default parameters. For the benchmark, we retained all PSMs with Percolator q value < 0.01. We identified 13 MGF files with fewer than 100 accepted PSMs, and we eliminated all of these PSMs from the benchmark. At this point in the processing pipeline, the dataset contains 2,898,611 annotated spectra (PSMs) drawn from 343 RAW files and associated with 168,422 distinct peptides.
Avoiding train/test leakage
To avoid train/test leakage, we post-processed the PSMs to eliminate peptides that are shared between species. Among the 168,422 distinct peptides, we identified 4121 (2.4%) that occur in more than one species. For each such peptide, we selected one of the associated species at random and then eliminated all PSMs containing that peptide in other species. Note that when identifying shared peptides between species, we considered all modified forms of a given peptide sequence to be the same, and we converted all isoleucines to leucines. Hence, if a given peptide appears in more than one species, then that peptide, including all its modified forms, is randomly assigned to a single species and eliminated from the others. The final, non-redundant benchmark dataset (“main”) consists of 2,838,117 PSMs corresponding to 168,422 distinct peptides.
Balancing the benchmark
At this stage, the benchmark was quite imbalanced, in the sense that some species had a much larger number of associated PSMs. We therefore used a random downsampling procedure to produce a benchmark that is more evenly balanced across species. Among the nine species, the one with the fewest PSMs is Mus musculus, with 25,522. Downsampling all of the other eight species to have 25,000 PSMs would reduce the size of the dataset from 2.8 million PSMs to 225,000—a reduction of 92%. To avoid producing such a small dataset, we therefore opted to downsample each dataset to approximately 100,000 PSMs. This approach yields a slight imbalance, because three species have fewer than 100,000 PSMs (44,555 for H. sapiens and 82,514 for Candidatus endoloripes), while retaining a larger percentage of the original data. Our downsampling procedure involved randomly permuting the order of the MGF files for each species and then selecting the files in order until at least 100,000 PSMs have been accepted. The final, balanced benchmark dataset is approximately one quarter the size of the main benchmark, consisting of 779,879 PSMs from 133,232 distinct peptides.
Data Records
The dataset35 contains files resulting from various steps in the generation of the benchmark:
-
Spectrum files in MGF format, produced by ThermoRawFileParser.
-
Reference proteome files in FASTA format, downloaded from UniProt.
-
Search results files for both targets and decoys, in tab-delimited format, produced by Tide.
-
PSM-level Percolator results files for targets, in tab-delimited format.
-
Annotated MGF and corresponding mzSpecLib36 files for both versions of the benchmark (main and balanced).
Also included are log files for the steps of the analysis pipeline carried out using Crux37 (Tide indexing, Tide search, and Percolator). The data is available at https://doi.org/10.5281/zenodo.13685813.
Technical Validation
Data quality and interpretability varies dramatically from study to study, due to differences in sample type, sample preparation protocols, chromatography and instrument settings, and database size. To assess the overall rate of successful identification of spectra in each data set, we plotted the number of accepted PSMs as a function of PSM-level FDR threshold (Fig. 1a). As is typical in proteomics database search, the curves go up rapidly before leaving the y-axis, corresponding to the many spectra with highly confident peptide assignments. To better understand the relative quality of the datasets, we also computed the proportion of spectra that were accepted at 1% PSM-level FDR per species (Fig. 1b). Here we observe that some datasets yield much higher rates of accepted PSMs than others, up to 39.7% for Saccharomyces cerevisiae and down to 3.6% for Candidatus endoloripes. Despite this large variance in the rate of accepted PSMs, characterizing the proportion of the total peak intensities that is explained by matched b- and y-ions (Fig. 1c) suggests that the quality of the accepted PSMs is high. Notably, the proportion of matched b- and y-ions does not appear to be strongly correlated with the rate of accepted PSMs per species.

Validation of the benchmarks. (a) Each series indicates, for a given species, the number of accepted PSMs as a function of PSM-level FDR. (b) The bar plot indicates the proportion of spectra that were accepted at 1% PSM-level FDR per species. (c) Each histogram shows, for one species, the distribution of the proportion of total ion current that is matched by b- or y-ions per accepted PSM, using a matching tolerance of 0.05 m/z.
Code availability
All code required to generate the various benchmarks and to produce the figures in this manuscript is available with an Apache license at https://github.com/Noble-Lab/multi-species-benchmark, with a snapshot of the repository stored at https://doi.org/10.5281/zenodo.12926326.
References
Sakurai, T., Matsuo, T., Matsuda, H. & Katakuse, I. Paas 3: A computer program to determine probable sequence of peptides from mass spectrometric data. Biomedical Mass Spectrometry 11, 396–399 (1984).
Tran, N. H., Zhang, X., Xin, L., Shan, B. & Li, M. De novo peptide sequencing by deep learning. Proceedings of the National Academy of Sciences of the United States of America 31, 8247–8252 (2017).
Bittremieux, W. et al. Deep learning methods for de novo peptide sequencing. chemRxiv https://chemrxiv.org/engage/chemrxiv/article-details/665189f821291e5d1d3838eb (2024).
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods 4, 207–214 (2007).
Karunratanakul, K., Tang, H.-Y., Speicher, D. W., Chuangsuwanich, E. & Sriswasdi, S. Uncovering thousands of new peptides with sequence-mask-search hybrid de novo peptide sequencing framework. Molecular and Cellular Proteomics 18, 2478–2491 (2019).
Eloff, K. et al. De novo peptide sequencing with InstaNovo: Accurate, database-free peptide identification for large scale proteomics experiments. bioRxiv 2023–08 (2023).
Liu, K., Ye, Y., Li, S. & Tang, H. Accurate de novo peptide sequencing using fully convolutional neural networks. Nature Communications 14, 7974 (2023).
Yang, T. et al. Introducing π-HelixNovo for practical large-scale de novo peptide sequencing. Briefings in Bioinformatics 25, bbae021 (2024).
Yang, Y. et al. DPST: de novo peptide sequencing with amino-acid-aware transformers. arXiv preprint arXiv:2203.13132 (2022).
Yang, H., Chi, H., Zeng, W., Zhou, W. & He, S. pNovo 3: precise de novo peptide sequencing using a learning-to-rank framework. Bioinformatics 35, i83–i90 (2019).
Xia, J. et al. AdaNovo: Adaptive de novo peptide sequencing with conditional mutual information. arXiv:2043.07013v1 (2024).
Wu, S., Luan, Z., Fu, Z., Wang, Q. & Guo, T. BiATNovo: A self-attention based bidirectional peptide sequencing method. bioRxiv 2023–05 (2023).
Qiao, R. et al. Computationally instrument-resolution-independent de novo peptide sequencing for high-resolution devices. Nature Machine Intelligence 3, 420–425 (2021).
Liu, Z. & Zhao, C. A residual network for de novo peptide sequencing with attention mechanism. In 2020 16th International Conference on Control, Automation, Robotics and Vision (ICARCV), 1165–1170 (IEEE, 2020).
Lee, S. & Kim, H. Bidirectional de novo peptide sequencing using a transformer model. PLOS Computational Biology 20, e1011892 (2024).
Klaproth-Andrade, D. et al. Deep learning-driven fragment ion series classification enables highly precise and sensitive de novo peptide sequencing. Nature Communications 15, 151 (2024).
Jin, Z. et al. ContraNovo: A contrastive learning approach to enhance de novo peptide sequencing. Proceedings of the AAAI Conference on Artificial Intelligence. 38(1), 144–152 (2024).
Ge, C. et al. DePS: an improved deep learning model for de novo peptide sequencing. arXiv preprint arXiv:2203.08820 (2022).
Ebrahimi, S. & Guo, X. Transformer-based de novo peptide sequencing for data-independent acquisition mass spectrometry. IEEE 23rd International Conference on Bioinformatics and Bioengineering (BIBE), Dayton, OH, USA, 2023, pp. 28–35 (2023).
Yilmaz, M., Fondrie, W. E., Bittremieux, W., Oh, S. & Noble, W. S. De novo mass spectrometry peptide sequencing with a transformer model. In Proceedings of the International Conference on Machine Learning, 25514–25522 (2022).
Yilmaz, M. et al. Sequence-to-sequence translation from mass spectra to peptides with a transformer model. Nature Communications (2024). In press.
Diament, B. & Noble, W. S. Faster SEQUEST searching for peptide identification from tandem mass spectra. Journal of Proteome Research 10, 3871–3879 (2011).
Käll, L., Canterbury, J. D., Weston, J., Noble, W. S. & MacCoss, M. J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nature Methods 4, 923–925 (2007).
Paiva, A. L., Oliveira, J. T., de Souza, G. A. & Vasconcelos, I. M. Label-free proteomic reveals that cowpea severe mosaic virus transiently suppresses the host leaf protein accumulation during the compatible interaction with cowpea (Vignaunguiculata[L.]Walp.). Journal of Proteome Research 15, 4208–4220 (2016).
Nevo, N. et al. Impact of cystinosin glycosylation on protein stability by differential dynamic stable isotope labeling by amino acids in cell culture (SILAC). Molecular and Cellular Proteomics 16, 456–468 (2017).
Cassidy, L., Prasse, D., Linke, D., Schmitz, R. A. & Tholey, A. Combination of bottom-up 2D-LC-MS and semi-top-down GelFree-LC-MS enhances coverage of proteome and low molecular weight short open reading frame encoded peptides of the archaeon Methanosarcinamazei. Journal of Proteome Research 15, 3773–3783 (2016).
Reuß, D. R. et al. Large-scale reduction of the Bacillus subtilis genome: consequences for the transcriptional network, resource allocation, and metabolism. Genome Research 27, 289–299 (2017).
Petersen, J. M. et al. Chemosynthetic symbionts of marine invertebrate animals are capable of nitrogen fixation. Nature microbiology 2, 1–11 (2016).
Mata, C. I. et al. In-depth characterization of the tomato fruit pericarp proteome. Proteomics 17, 1600406 (2017).
Seidel, G. et al. Quantitative global proteomics of yeast PBP1 deletion mutants and their stress responses identifies glucose metabolism, mitochondrial, and stress granule changes. Journal of Proteome Research 16, 504–515 (2017).
Hu, H. et al. Proteome analysis of the hemolymph, mushroom body, and antenna provides novel insight into honeybee resistance against varroa infestation. Journal of Proteome Research 15, 2841–2854 (2016).
Cypryk, W., Lorey, M., Puustinen, A., Nyman, T. A. & Matikainen, S. Proteomic and bioinformatic characterization of extracellular vesicles released from human macrophages upon influenza a virus infection. Journal of Proteome Research 16, 217–227 (2017).
Hulstaert, N. et al. ThermoRawFileParser: modular, scalable and cross-platform raw file conversion. Journal of Proteome Research 19, 537–542 (2020).
Sulimov, P. & Kertész-Farkas, A. Tailor: A nonparametric and rapid score calibration method for database search-based peptide identification in shotgun proteomics. Journal of Proteome Research 19, 1481–1490 (2020).
Wen, B. & Noble, W. A multi-species benchmark for training and validating large scale mass spectrometry proteomics machine learning models. Zenodo https://doi.org/10.5281/zenodo.13685813 (2024).
Klein, J. et al. The Proteomics Standards Initiative standardized formats for spectral libraries and fragment ion peak annotations: mzSpecLib and mzPAF. chemRxiv https://doi.org/10.26434/chemrxiv-2024-p71gj (2024).
Park, C. Y., Klammer, A. A., Käll, L., MacCoss, M. P. & Noble, W. S. Rapid and accurate peptide identification from tandem mass spectra. Journal of Proteome Research 7, 3022–3027 (2008).
Acknowledgements
This work was supported by NSF award 2245300.
Author information
Authors and Affiliations
Contributions
W.S.N. prepared the dataset, performed the validation experiments, and wrote the manuscript. B.W. edited the manuscript and wrote the code to convert MGF to mzSpecLib.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wen, B., Noble, W.S. A multi-species benchmark for training and validating mass spectrometry proteomics machine learning models. Sci Data 11, 1207 (2024). https://doi.org/10.1038/s41597-024-04068-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-04068-4
