
Abstract
The accumulation of large datasets by the scientific community has surpassed the capacity of traditional processing methods, underscoring the critical need for innovative and efficient algorithms capable of navigating through extensive existing experimental data. Addressing this challenge, our study introduces a machine learning (ML)-powered search engine specifically tailored for analyzing tera-scale high-resolution mass spectrometry (HRMS) data. This engine harnesses a novel isotope-distribution-centric search algorithm augmented by two synergistic ML models, assisting with the discovery of hitherto unknown chemical reactions. This methodology enables the rigorous investigation of existing data, thus providing efficient support for chemical hypotheses while reducing the need for conducting additional experiments. Moreover, we extend this approach with baseline methods for automated reaction hypothesis generation. In its practical validation, our approach successfully identified several reactions, unveiling previously undescribed transformations. Among these, the heterocycle-vinyl coupling process within the Mizoroki-Heck reaction stands out, highlighting the capability of the engine to elucidate complex chemical phenomena.
Similar content being viewed by others

Introduction
The role of experiments is crucial to confirming hypotheses and making discoveries in chemical science. However, the procedures used may take a long time due to limitations of the method, costs of reagents/catalysts, difficulties in waste handling, operational delays, and a considerable amount and complexity of the analyzed data. Therefore, two strategies are primarily used to decrease time and human resources for performing experiments: automation of data acquisition (e.g., in automated chemical syntheses1,2,3, in mass spectrometry-based proteomics4,5 or high-throughput microscopy6,7) and automation of data interpretation (chemical space exploration8,9,10, NMR data11,12,13, and mass spectrometry—MS—data14,15,16,17,18).
However, one can think about a third feasible strategy, to use previous results (already existing data) for hypothesis testing, thus reducing the number of experiments. The fundamental limitations of the strategy include the possible lack of accessible scientific data and its management with FAIR19 (findable, accessible, interoperable, and reusable) principles. This disadvantage can be eliminated by maintaining common open databases of experimental data20,21 with detailed descriptions of experiments within the laboratory or by using web applications that enable remote collaborative research in a shared analysis environment22. Another important disadvantage is the lack of dedicated software for the implementation and deployment of chemically efficient algorithms to search/extract data.
In a typical organic synthesis workflow, chemists select particular experimental conditions for the optimization of a reaction to achieve the maximal outcome for the desired product (Figure S1, as an example). Next, the reaction and sample preparation are carried out, followed by detection and characterization of the chemical compositions of the studied system using an appropriate analytical system (Fig. 1a). High-resolution mass spectrometry (HRMS) is an excellent, very often used method to execute this strategy due to its high speed of analysis, sensitivity and ease of data accumulation23. HRMS is widely used in analytical chemistry24, organic25,26,27 and inorganic chemistry28, proteomics29, metabolomics30, petroleomics31, metal complex catalysis32,33,34,35,36, organocatalysis37, polymer science38, and material science39, among many other directions.

a A commonly used standard experimental workflow for reaction optimization with limited manual human interpretation of results and incomplete reaction space investigation. As an example, we highlight analysis of high-resolution mass spectrometry (HRMS) data that contains significant amount of information about reaction mixture. b “Experimentation in the past” concept is achieved using a machine learning (ML)-powered search engine with the discovery of new pathways in Mizoroki-Heck and hydrothiolation reactions in a large amount of already existing data. The search consists of hypothesis generation using various methods (manual, automated fragment-based, and enabled by large language models, LLM), and search itself, which uses already existing HRMS data.
Within a routine research pipeline, HRMS-equipped laboratories produce mass spectral data every day. During a relatively short period of time, data storage may contain tens of thousands of recorded files. Some spectra weigh several gigabytes (e.g., reaction monitoring spectra at high resolution), in overall, leading to terabytes of recorded information being stored on computer drives. Currently, manual analysis connects experiments with MS data (Fig. 1a). This approach imposes serious limitations associated with incomplete interpretation coverage of the analyzed data due to human factors. Mainly, only the desired product and a few known byproducts are looked at, leaving most MS signals unattended. Within a few years of experimental work in the laboratory, terabytes of data are accumulated and stored.
Thus, many new chemical products have already been accessed, recorded, and stored with HRMS but remain undiscovered. Therefore, the development of methods that can screen terabyte-scale databases and collect molecular patterns opens the way for cost-efficient and environmentally friendly chemistry discoveries while operating on existing stored data with no new experiments needed.
In this study, we demonstrate that in the case of mass spectrometry, data analysis can be implemented as a search engine with automated ion detection algorithms (Fig. 1b). An ultimate digital tool for accelerated discovery would allow searching for automatically generated ion candidates in a vast array of existing complex mass spectra with high accuracy in a reasonable amount of time and hardware resources. The algorithm can not only search known/existing products but also comprehensively search for unknown products, transformation pathways, contaminants, etc. The proposed approach makes already existing data a perfect source for reaction discovery in advantageously Green and Sustainable ways (no chemicals are consumed; no waste). Importantly, even though we only reveal the presence of ions with specific molecular formulas, the user may supplement the study further by designing experiments to verify the structure manually using either orthogonal methods such as NMR (Nuclear Magnetic Resonance) spectroscopy, or by obtaining tandem mass spectrometry (MS/MS) data. In our examples, we show how this can be done.
In the case of this approach to achieve this aim, a powerful algorithm to search compounds in large-scale MS data is a key requirement. To date, search algorithms for complex (with more than one compound in the spectrum) mass spectrometry data have been actively used, mainly in metabolomics40,41,42 and proteomics43,44,45,46,47,48 studies. The search is primarily based on matching peaks in the experimental MS/MS spectrum with peaks in the theoretical spectrum obtained from the peptide sequence49. There are also examples of structural annotation and exploring genomics50 and metabolomics51,52,53,54,55 datasets with MS/MS data. FastEI56 software uses Word2vec to transform the electron ionization spectra into embeddings with further large-scale spectrum matching. However, typical workflows have limited applicability due to incomplete chemical space coverage, i.e., the narrow application scope of such engines. Moreover, although already implemented in some packages57,58, we would like to stress the importance of isotopic distribution59 patterns, which leads to false detections (see SI Section S2 for a relationship between the isotopic distribution information and false positive rate).
Furthermore, annotated training data inaccessibility in supervised machine learning (ML) for mass spectrometry continues to be a major bottleneck due to the lack of human resources, time for labeling data, and high dimensionality of mass spectra. Model learning requires up to several thousand labeled ions to achieve good performance. Synthetic data can be used to solve this problem (see SI Section S3 for details on simulated spectra use). Artificially generated spectra were previously used in ML model training and showed their applicability in MS tasks: atomic pattern recognition60, deisotoping15, and the “inverse problem” of molecular identification15. MS spectra augmentation techniques are also widely studied61,62.
Considering the disadvantages above, this work proposes an approach for searching in large stored arrays of mass spectral data with a focus on reaction discovery (Fig. 1b). The models were trained on synthetic data. The key contribution of the work is the development of a search engine, called MEDUSA Search, that allows the finding of ion isotopic distributions in a tera-scale database (in our case, more than 8 TB of 22 000 spectra) of multicomponent HRMS spectra with different resolutions in an acceptable time (see the “Reaction discovery approach” section for dataset description). The engine is able to confirm basic hypotheses of the presence of ions of interest in a wide range of applications (i.e., support all possible ion formulas with different charges). As an illustrative example, we applied the developed algorithm to HRMS data accumulated by many research groups studying a large scope of diverse chemical transformations, including the well-known and industrially relevant Mizoroki–Heck reaction (see SI Section S1). The data were collected over a few years and remained abandoned. We demonstrate that new transformations may be discovered upon automated search of archived data. Since this reaction has been widely known and studied numerous times previously by various scientific groups, it is important to demonstrate the advantage of the developed computational approach to reveal “surprising” transformations, which have been overlooked in manual analysis for years.
In this way, the concept of “experimentation in the past”, an approach to research when a researcher uses experimental data made earlier instead of conducting a new experiment, was achieved with the discovery of novel catalyst transformation pathways in cross-coupling and hydrogenation reactions. Importantly, data reuse and repurposing are already common in fields such as proteomics and metabolomics. However, research related to organic chemistry is quite limited21.
Results and discussion
Overview of the search engine
To proceed with the reaction discovery workflow, it is first necessary to develop a search engine, which underlies the proposed approach. The Machine-Learning-Powered search pipeline developed in MEDUSA Search consists of five overall steps, as illustrated in Fig. 2 and described in the text below. The multilevel architecture of the system is inspired by existing web search engines and is crucial to achieve satisfactory search speeds (see SI Section S4 for search speed tests).

First, the engine takes as input molecular formulas and charges of searched ions. They can be derived from the reaction system using hypothesis generation method (through fragment-based or large language model, LLM, guided approach) or defined manually (A). Then, it searches all spectra files that contain the two most abundant isotopologues’ peaks of each input ion (B). The peak is represented by its mass-to-charge ratio—m/z. These spectra files are called the candidates. Cosine distance threshold is calculated for them (C1). Then, an algorithm that searches for the isotopic distribution by input formula within a single spectrum is performed for all candidate mass spectra (C2). Additional machine learning (ML) models attempt to decrease the number of false positive search answers (C3).
Importantly, all the ML models were trained without the use of large number of annotated mass spectra. This was done by generating synthetic MS data with the construction of isotopic distribution patterns from molecular formulas and the following data augmentation to simulate measurement errors of the instrument (see SI Section S3 for details).
Before searching, we need to generate a list of hypothesis reaction pathways on the basis of our prior knowledge about the reaction system (Fig. 2, step A). Here, we design this system around breakable bonds and the recombination of corresponding fragments. If a user understands which bonds may break and form, they may supply individual fragments that will be automatically combined to create a query ion. However, we also allow BRICS63 fragmentation or the use of multimodal LLMs to perform this fragmentation (see Section S5 for examples of generated hypotheses). The development of new hypothesis generation methods is an open problem, and any new work in the field can be easily integrated into this system.
Input information about the chemical formula and charge allows us to calculate the theoretical “isotopic pattern” of the ion. The two most abundant isotopologue peaks are searched in inverted indexes (see SI Section S6.1 for details) with an accuracy of 0.001 m/z (Fig. 2, step B). Mass spectra that contain these peaks are called candidates. The following isotopic distribution search will be performed on them.
After a coarse spectra search, an isotopic distribution search of the query ion is performed for each candidate spectrum. This step includes 1) initial ion presence threshold estimation; 2) in-spectrum isotopic distribution search; and 3) filtering false positive matches. Descriptions of each step are given below.
The in-spectrum isotopic distribution search algorithm returns the cosine distance as a metric of similarity between theoretical and matched isotopic distributions (see SI Section S6.2 for algorithm details). The automatic decision of whether there is an ion in the spectrum or not depends on the estimated maximum cosine distance (i.e., ion presence threshold), which depends on the formula of the query ion (see Figure S8d for the threshold/formula relationship). A machine learning (ML) regression model is implemented (Fig. 2, step C1) to determine the ion presence threshold with the input ion formula (see SI Sections S3, S6.3, and S6.4 for data generation, data encoding and performance evaluation, and hyperparameter tuning, respectively).
The in-spectrum isotopic distribution search algorithm (Fig. 2, step C2) matches peaks from the experimental candidate mass spectrum with peaks from the theoretical isotopic distribution; at each step, the cosine distance is calculated, which allows the selection of the most similar peaks. If no peak is found, it is replaced with a peak with an intensity equal to the median of the noise. If the final cosine distance is less than the ion presence threshold, estimated on Step C1, the ion is considered to be found (for more details, see SI Section S6.2).
An additional ML classifier (Fig. 2, step C3) detects false positive ion presence verification with information about neighboring peaks (see SI Section S3 for training data generation). This problem usually appears as selecting the searched distribution as a part of another distribution. One of the most prominent examples starts with M + 1, while M is also present (see SI Section S6.5 for performance and interpretability studies; Section S6.6 for hyperparameter tuning; Section S7 for false positive examples).
To facilitate the work with the search engine, the Command Line Interface (CLI) was developed using the Click Python package (see SI Section S8 for more information).
Reaction discovery approach
Having various hypotheses about the course of possible new reactions, it is necessary to cover as much chemical space as possible. In this work, combinatorial generation of molecular formulas of proposed products (i.e., rule-based generation of molecular formulas with unique structures but different substituents) was performed to connect reaction discovery with the automated mass spectral ion search in already existing data. The FAIR description data from previous experiments (Fig. 1b) are also essential for validating the search results in practice.
The search for novel reactions included more than 20,000 mass spectra without any prior knowledge of their composition (Fig. 3b). The search procedure placed no limitations on the filename, the researcher’s name, who recorded the spectrum or any other aspect of decreasing the search space. One commonly used method for visualizing complex data is through the application of the t-SNE dimensionality reduction technique64. To demonstrate the high diversity of the archived data set, two t-SNE plots were created. As shown in Fig. 3a, the compounds registered in the analyzed mass spectra cover the chemical space well. In Fig. 3b, each point represents a spectrum, and similar mass spectra are located close to each other on the plot (see SI Section S9 for t-SNE plot generation details and to see the enlarged version of the t-SNE map). It is evident that various workers record diverse spectra that contrast from one another. Moreover, one can also see common projects, where multiple people record similar spectra. Instrument operator C has the most widespread distribution of mass spectra, which matches their primary role — recording data for sample drop-off service for the entire institute.

a t-distributed stochastic neighbor embedding (t-SNE) map of chemical structures encoded with Morgan fingerprints. Molecules were collected via random sampling from the PubChem database and from compounds that were registered in the mass spectra used in the research. Source data are provided as a Source Data file; b t-SNE map of the archived MS data used in the research (see Figure S20 for the enlarged version). Each point represents a unique mass spectrum. Different colors indicate instrument operators (coded by letters) who recorded mass spectra. Operator C registers mass spectra for the entire institute. Source data are provided as a Source Data file; c Functional groups and ligands, which were used in the generation process; NHC—N-heterocyclic carbene, Ar — aryl group, Nu — nucleophile, EWG — electron-withdrawing group. d The generation of ion formulas involves a complete enumeration of all functional groups and ligands for each core; e Bar chart illustrating the number of detected ions, categorized by the type of transformation. Source data are provided as a Source Data file.
The discovery of intermediates in organic reactions is essential to understand the mechanism and propose new strategies for reaction design and optimization. Electrospray ionization mass spectrometry (ESI-MS) is widely used in these studies65,66,67,68. It is also used as a method to characterize synthetized products69,70. To demonstrate the applicability of the developed search engine, it was used to find new transformation pathways in Pd/NHC-catalyzed (NHC = N-heterocyclic carbene) reactions71 with the combined generation of ion formulas (Fig. 3c). For each formula component (functional group or NHC-ligand), which is contained in one of the 13 analyzed structural cores (Fig. 3d), the molecular formula was calculated. The total number of generated ion formulas was 520, and 400 out of which had unique mass. Importantly, HRMS without fragmentation techniques can provide information only about molecular formulas; thus, structural isomers cannot be distinguished.
Once the hypothesis set was generated, the method was applied to attempt to verify them using previously collected data and retrieved laboratory notebooks. A search pipeline (Fig. 2) was run for each of 520 generated ions through the entire tera-scale HRMS database (see SI Section S9 for data set information), with a total computational time of 3–4 days (8–11 minutes per ion). As a result, the engine detected many isotopic distribution patterns. However, most of the search engine answers could not be validated because of the lack of FAIR description data needed for recognition of the initial composition of the reaction mixture. Nevertheless, some samples were checked via laboratory notebooks. The collected results (see SI Section S10 for MS spectra) included the following:
-
1.
The presence of corresponding azolium salts (m/z 147) in all reactions associated with M/NHC catalysis72 (Fig. 4a);
Fig. 4: The search engine enables the discovery of novel products in old data (HRMS spectra of previously unknown reaction products are indicated by dotted purple arrows). 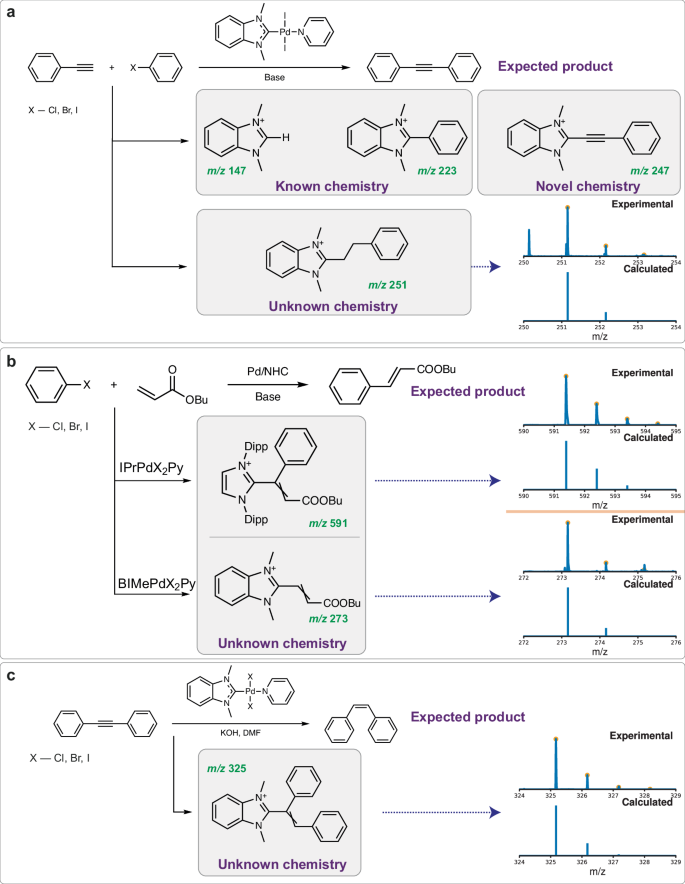
a MEDUSA Search has registered widely known H-NHC and Ph-NHC ions, as well as a newly discovered [NHC-ethynyl]+ ion in a Pd/NHC-catalyzed Sonogashira reaction mixture. The isotopic distribution-informed search process allows the detection of previously unknown ethyl-NHC products; b MEDUSA Search registered unknown vinyl-NHC fragments in the Mizoroki-Heck reaction (Dipp – 1,3-diisopropylphenyl, Py – pyridine); c MEDUSA Search registered phenyl-substituted vinyl-NHC ions in the Pd-PEPPSI (Pyridine-Enhanced Precatalyst Preparation Stabilization and Initiation) catalytic hydrogenation reaction.
-
2.
The presence of known [phenyl-NHC]+ ions73 (m/z 223) in cross-coupling reactions (Fig. 4a);
-
3.
The presence of a recently discovered [ethynyl-NHC]+ ion74 (m/z 247) in the Sonogashira reaction (Fig. 4a);
-
4.
The presence of an unknown [ethyl-NHC]+ ion (m/z 251) in the Sonogashira reaction (Fig. 4a);
-
5.
The presence of unknown [vinyl-NHC]+ (m/z 273) and [vinyl-phenyl-NHC]+ (m/z 591) ions75 in the Pd/NHC-catalyzed Mizoroki-Heck reaction in the spectra recorded by different researchers in different years (Fig. 4b);
-
6.
The presence of an unknown [vinyl-NHC]+ ion (m/z 325) in Pd/NHC catalyzed the hydrogenation reaction (Fig. 4c).

Figure 3e presents statistics regarding the number of ions detected during the search procedure. All of these ions had unique masses. The preferred type of transformation is phenyl–NHC coupling. Compared with other types of transformations, vinyl-NHC coupling is infrequent. The obtained results are correlated with quantum chemical study of transformation pathways (see SI Section S15 for full information about quantum chemical study). Notably, the validation of the search results using laboratory notebooks was only possible for a limited number of mass spectra. For most ions, it is unclear in which reactions they were discovered and if they truly correspond to the assumed structural formula. Thus, further experimental validation is needed (Fig. 5).

a The formation of [BIMe(CH)2COOBu]+ ion was proven with ESI-HRMS; b the formation of [IPrCHC(Ph)COOBu]+ ion was proven with ESI-HRMS; c MS/MS spectrum of [IPrCHC(Ph)COOBu]+ ion; d vinyl-NHC and ethyl-NHC reaction products (Dipp – 1,3-diisopropylphenyl, Mes - mesityl).
In addition to the main search procedure employed for the identification of previously unknown products in Pd/NHC-catalyzed reactions, an alternative example of search engine capabilities was pursued through the discovery of nickel-catalyzed hydrothiolation reaction side products76 (see Section S10.2 for details).
Experimental validation
The formation of the catalyst transformation products shown in Fig. 3d is strongly related to the corresponding reaction mechanism. Previously, we conducted several Mizoroki-Heck and cross-coupling reactions (e.g., Sonogashira, Suzuki, Buchwald-Hartwig, etc.) catalyzed by Pd/NHC complexes with different NHC ligands and halogen substituents. During the investigation of the reaction mechanisms via ESI-MS spectra of the reaction mixtures, the coupling products [NHC-H]+, [NHC-Ph]+, [NHC-O]+, and [NHC-N]+ were found. On the basis of these observations, the key role of R-NHC coupling and M-NHC bond cleavage in the evolution of M/NHC complexes under catalytic reaction conditions was revealed73. The formation of catalytically active molecular M/NHC catalysts and “NHC-free” cocktail-type catalysts, including the formation of H-NHC salts77 and O-NHC coupling78, was described first in terms of the number of C‒C coupling reactions.
In the Sonogashira reaction, the previously unknown product of the ethynyl-NHC coupling product was isolated, and possible reaction pathways were described74. The ethynyl-NHC coupling product is very reactive and may undergo various transformations. Using the described approach for hydrogenated derivatives of the product revealed the presence of the [NHC-(CH2)2-Ph]+ product in the ESI-MS spectra of the Sonogashira reaction mixtures (Fig. 4a). Presumably, the process occurs via a kind of transfer hydrogenation reaction.
Similar to the discovery of ethynyl-NHC and aryl-NHC coupling products, we envisioned the possibility of the formation of two different vinyl-NHC coupling products (Fig. 4b) before and after the insertion step in the Mizoroki–Heck reaction. Both products were observed in the experimental reaction mixtures. Here, we also aimed to perform experimental validation of the observed reaction. To do that, original lab notebooks were retrieved, and corresponding experiments were found. Mass spectrometry analysis of the reaction mixtures of the Mizoroki–Heck reaction between p-methoxyiodobenzene and butyl acrylate, catalyzed by the Pd/NHC complex [BIMePh]+[BIMePdI3]-, revealed the formation of [BIMe(CH)2COOBu]+ (Fig. 5a). The molecular formula was confirmed with ultrahigh-resolution mass spectrometry. The experiment involving the formation of [IPrCHC(Ph)COOBu]+ was a mercury test for distinguishing between homogenous and heterogeneous catalysis. We excluded mercury to avoid interference with reactive species79 and kept other conditions as in the original experiment. The molecular formula was also confirmed with ultrahigh-resolution mass spectrometry (Fig. 5b); the chemical structure was verified with MS/MS experiment (Fig. 5c).
We also conducted experiments using different NHC ligands (see SI Section S11 for experimental details). The possibility of vinyl-NHC coupling in the process of Pd/NHC transformation under the Mizoroki–Heck reaction was tested with five different NHC palladium complexes, as illustrated in Figure S35. We used Pd complexes with different co-ligands to prove the generality of vinyl-NHC coupling under catalytic conditions. The scope is summarized in Fig. 5d. The vinyl-NHC coupling products were registered, confirming the proposed reaction (see SI Section S12.1 “Ultrahigh-resolution mass spectra”, Figures S37–S45). The vinyl-NHC product was found in all studied cases, independent of the ligand in the complex, with an ultrasmall definition error for all of them. Along with vinyl-NHC, ethyl-NHC was also detected in all the investigated reaction mixtures, for (BIMe)PdI2Py, (SIMes)PdCl(allyl), and (PIPr)PdCl(allyl), with very low errors m/z errors of less than 0.3 ppm and low errors of less than 1 ppm in the case of the (IMes)PdCl(allyl) and (SIPr)PdCl(allyl) complexes. In all MS experiments, we set configurations to prevent transformations during the recording of mass spectra (see SI Section S13 for more information). Pressure sample infusion ESI-MS reaction monitoring for the discussed vinyl–NHC coupling process was also performed to confirm that ions can be observed across multiple modalities of the reaction data collection (see SI Section S12.2).
Finally, in the transfer hydrogenation reaction (Fig. 4c), another type of ethynyl-NHC coupling could be observed. Indeed, the search revealed the formation of the corresponding product. The described transformation sheds light on the dynamic nature of catalytic systems and opens opportunities for the development of Pd-catalyzed imidazole ring functionalization reactions.
To gain insights into the mechanisms of the discovered transformations and additionally confirm their feasibility from a theoretical point of view, a DFT quantum chemical study was performed, which confirmed the reaction channel for the vinyl-NHC coupling and identified the possibility of this newly discovered reaction occurring (see SI Section S15 for the computational results).
In this work, a robust ML-based computational engine for reaction discovery was developed. First, we start with automated methods for compound hypothesis generation. The selected candidates are then passed to the search engine. The combination of an isotopic distribution-based algorithm with two additional machine learning models made it possible to reduce false positive ion detection, which was crucial to increase search performance in databases of various objects of study. The steps of the search workflow were optimized, synthetically and experimentally validated. The interpretability of the models allowed us to obtain an understanding of how these models behave. A reduction in the ion search space with the account of isotopic distribution patterns proved the advantage of the isotope-distribution-centric approach.
The ability of the engine to use a wide range of ions with different compositions showed the excellent applicability of the system. An ion search can be performed on all MS instruments with a resolution that allows the observation of the isotopic distribution. A combination of the developed system with other computational techniques (e.g., algorithms for the prediction of ion fragments by structural formula or peptide sequence, different adduct calculators) can become a powerful analytical tool for comprehensive screening, which is vital for accelerated discovery in various scientific fields.
Moreover, even though the presence of FAIR data description is a major requirement of our approach, users can perform multiple queries to reduce the false positive rate of the system. For example, searching not only for the expected product but also for the corresponding reagents will significantly narrow down the scope for experimentation. However, ultimately, we consider this work an important step in raising awareness of how critical proper data collection and description are.
As an example, the developed search engine allowed the discovery of previously unknown M/NHC-catalyzed reaction byproducts, saving the resources needed to confirm hypotheses with the concept of “Experimentation in the past”. In this approach, two degrees of novelty were achieved:
-
1.
Reaction pathway novelty — the reactions unexpected for this particular process but known and reported for other catalytic processes. In this work, we showed the formation of H-NHC salts and ethynyl-NHC coupling products. For these processes, the findings of our computational approach can be validated by comparison with those of other methods, including NMR spectroscopy and single-crystal X-ray analysis. These findings are important to connect the studied reaction with other processes and enhance catalyst development principles with relationships documented for other processes.
-
2.
Totally new reactions/products (never reported before). Here, we demonstrated the possibility of a vinyl–NHC coupling process in the Mizoroki–Heck reaction. [BIMe(CH)2COOn-Bu]+[X]- and [IPrCHC(Ph)COOn-Bu]+[X]- are new compounds that have never been reported and are absent from the SciFinder and Reaxys databases. The discovered transformation appears probable on the basis of general chemistry knowledge. Moreover, we observed various hydrogenated products that raise questions about the mechanism of their formation.
Here, we demonstrated that analysis of unused (old or abandoned) data with the developed computational algorithm can reveal pathways and reactions that were not described previously. Both degrees of novelty were verified, and the feasibility of the computationally revealed reactions was confirmed.
The discovered reaction pathways and reactions/products were rigorously verified by independent replication of experiments with different ligands and ultrahigh resolution MS measurements with m/z errors less than 1 ppm. To ensure that not only the molecular but also the structural formulas are correct, MS/MS ultrahigh resolution mass spectrometry analysis was performed. Mechanistic considerations and DFT study have increased confidence in the discovered reactions even more (see SI Section S15 for more details).
We plan to continue to work on the problem of interpretation of mass spectra and hope that in the future, automated analysis of MS data will become a major source of discoveries in chemistry.
Methods
General considerations
All starting materials, catalyst precursors and solvents were purchased from the commercial sources.
Mass spectra were measured using Bruker maXis instrument equipped with an electrospray ionization source (ESI) with Time-of-Flight (TOF) analyzer and spectra were recorded with m/z 50–1500 range. Capillary Voltage was set: for the positive ion mode to –4.5 kV, Spray Shield Offset was set to –0.5 kV. For calibration of the mass spectra a low-concentration tuning mix solution by Agilent Technologies was utilized. Nitrogen was applied as a nebulizer gas (0.4 bar) and dry gas (4.0 L × min−1, 250 °C). Bruker Data Analysis 5.1 software package was used.
Ultrahigh-resolution mass spectra were recorded on a Bruker solariX XR (ICR mass analyzer, a 15 T superconducting magnet) mass spectrometer equipped with an ESI source. The m/z scanning range was 100–1500. The number of scans was 256, with 8 M data points. External calibration of the mass scale was carried out using a sodium trifluoroacetate solution (0.1 mg/mL in a 1:1 acetonitrile/water mixture). The measurements were carried out in positive ion mode (+) (ground spray needle, 4500 V high-voltage capillary; HV end plate offset: –500 V). Nitrogen was used as the nebulizer gas (0.5 bar), and dry gas was used (4.0 L/min, 180 °C).
The chromatographic analysis was carried out on a chromatograph Agilent 1200 equipped with analytical column ZORBAX SB-C18 (2.1 × 50 mm); the size of the particles of the stationary phase 1.8 μm, mobile phase acetronitrile – 0.1% water solution of formic acid, 9:1, elution in the isocratic mode, flow rate 0.25 ml min-1, temperature 25 °C, the volume of injected sample 0.01 μl. The analyzed mixture was dissolved in acetonitrile (Merck, HPLC grade).
Experimental procedure for pressure sample infusion ESI-MS reaction monitoring (PSI-MS)
The mixture of Pd/NHC complexes (SIPr)PdCl(allyl) (0.015 mmol, 10 mg) and (PIPr)PdCl(allyl) (0.015 mmol, 10 mg) (see Figure S35 for structure details), n-butylacrylate (0.042 mmol, 6 μl) and dimethylformamide (2 mL) were mixed in Schlenk tube. Potassium tert-butoxide (0.06 mmol, 7 mg) was dissolved into isopropanol (600 μl) and the solution was added to the mixture. One side of Schlenk with reaction mixture was closed with a septum, the second side equipped a tap was connected with a «double» balloon with argon. An ion source of spectrometer was connected with the Schlenk by red PEEK capillary through the septum. The reaction monitoring has been carried out during 50 minutes at 140 °C. The spectra were acquired in positive ion mode and formation of the vinyl-NHC coupling products were observed at after 10 minutes of the start.
Computational details
DFT calculations were carried out in the Gaussian 16 (revision C.01) program80 via the PBE1PBE hybrid functional81. The 6-31 G** (for H, C, N, O, and Br atoms)82 and Def2TZVP (for Pd atom)83 basis sets were employed in the calculations. The empirical Grimme correction (GD3BJ)84 was used to take into account dispersion interactions. The influence of the solvent was taken into account via a polarizable continuum model (PCM)85. N,N-Dimethylformamide was used as the solvent. The geometry was optimized with subsequent calculation of vibrational frequencies and thermodynamic parameters for all the structures. All transition state structures had one negative vibrational frequency corresponding to the considered reaction path. The remaining structures had no imaginary vibrational frequencies and represented a minimum on the potential energy surface.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study have been deposited in Figshare86. The data repository also contains a 9 GB ZIP-archive of mass spectra with all mentioned found products. It is also enriched with additional reaction mass spectra and can be used to test the functionality of the developed search engine. The list of data in which the search was performed, and the results were not found, cannot be shared publicly due to confidentiality/IP considerations. The list of data can be accessed with instructions obtained from the authors upon request and being a subject of confidentiality/IP owners approval. Source data are provided with this paper.
Code availability
The code under GPL-3.0 license is available on GitHub at https://github.com/Ananikov-Lab/medusa-search. Additionally, it was deposited to Zenodo87.
References
Coley, C. W. et al. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science 365, 557–565 (2019).
Steiner, S. et al. Organic synthesis in a modular robotic system driven by a chemical programming language. Science 363, 144–144 (2019).
Minato, T. et al. Robotic stepwise synthesis of hetero-multinuclear metal oxo clusters as single-molecule magnets. J. Am. Chem. Soc. 143, 12809–12816 (2021).
Fu, Q. et al. Highly reproducible automated proteomics sample preparation workflow for quantitative mass spectrometry. J. Proteome Res. 17, 420–428 (2018).
Alexoviˇ, M., Sabo, J. & Longuespée, R. Automation of single-cell proteomic sample preparation. Proteomics 21, 1–11 (2021).
Wu, C., Huang, X., Cheng, J., Zhu, D. & Zhang, X. High-quality, high-throughput cryo-electron microscopy data collection via beam tilt and astigmatism-free beam-image shift. J. Struct. Biol. 208, 107396 (2019).
Schorb, M., Haberbosch, I., Hagen, W. J. H., Schwab, Y. & Mastronarde, D. N. Software tools for automated transmission electron microscopy. Nat. Methods 16, 471–477 (2019).
Caramelli, D. et al. Discovering new chemistry with an autonomous robotic platform driven by a reactivity-seeking neural network. ACS Cent. Sci. 7, 1821–1830 (2021).
Schwaller, P. et al. Mapping the space of chemical reactions using attention-based neural networks. Nat. Mach. Intell. 3, 144–152 (2021).
Schleinitz, J. et al. Machine learning yield prediction from NiCOlit, a small-size literature data set of nickel catalyzed C–O couplings. J. Am. Chem. Soc. 144, 14722–14730 (2022).
Howarth, A., Ermanis, K. & Goodman, J. M. DP4-AI automated NMR data analysis: straight from spectrometer to structure. Chem. Sci. 11, 4351–4359 (2020).
Yang, Z., Chakraborty, M. & White, A. D. Predicting chemical shifts with graph neural networks. Chem. Sci. 12, 10802–10809 (2021).
Atwi, R. et al. An automated framework for high-throughput predictions of NMR chemical shifts within liquid solutions. Nat. Comput Sci. 2, 112–122 (2022).
Gessulat, S. et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 16, 509–518 (2019).
Boiko, D. A., Kozlov, K. S., Burykina, J. V., Ilyushenkova, V. V. & Ananikov, V. P. Fully automated unconstrained analysis of high-resolution mass spectrometry data with machine learning. J. Am. Chem. Soc. 144, 14590–14606 (2022).
Phung, W., Bakalarski, C. E., Hinkle, T. B., Sandoval, W. & Marty, M. T. UniDec processing pipeline for rapid analysis of biotherapeutic mass spectrometry data. Anal. Chem. 95, 11491–11498 (2023).
Larson, E. J. et al. MASH Native: a unified solution for native top-down proteomics data processing. Bioinformatics 39, btad359 (2023).
Yunker, L. P. E., Donnecke, S., Ting, M., Yeung, D. & McIndoe, J. S. PythoMS: a python framework to simplify and assist in the processing and interpretation of mass spectrometric data. J. Chem. Inf. Model 59, 1295–1300 (2019).
Wilkinson, M. D. et al. The FAIR guiding principles for scientific data management and stewardship. Sci. Data 3, 160018 (2016).
Kearnes, S. M. et al. The Open Reaction Database. J. Am. Chem. Soc. 143, 18820–18826 (2021).
Jablonka, K. M., Patiny, L. & Smit, B. Making the collective knowledge of chemistry open and machine actionable. Nat. Chem. 14, 365–376 (2022).
Petras, D. et al. GNPS Dashboard: collaborative exploration of mass spectrometry data in the web browser. Nat. Methods 19, 134–136 (2022).
Burykina, J. V., Boiko, D. A., Ilyushenkova, V. V., Eremin, D. B. & Ananikov, V. P. Comprehensive mass spectrometric mapping of chemical compounds for the development of algorithms for machine learning and artificial intelligence. Dokl. Phys. Chem. 492, 51–56 (2020).
Meekel, N., Vughs, D., Béen, F. & Brunner, A. M. Online prioritization of toxic compounds in water samples through intelligent HRMS data acquisition. Anal. Chem. 93, 5071–5080 (2021).
Chen, M. & Dong, G. Copper-catalyzed desaturation of lactones, lactams, and ketones under ph-neutral conditions. J. Am. Chem. Soc. 141, 14889–14897 (2019).
Sahoo, H., Zhang, L., Cheng, J., Nishiura, M. & Hou, Z. Auto-tandem copper-catalyzed carboxylation of undirected alkenyl C–H Bonds with CO 2 by harnessing β-hydride elimination. J. Am. Chem. Soc. 144, 23585–23594 (2022).
Takimoto, M., Liu, M., Nishiura, M. & Hou, Z. Regioselective benzylic C–H Alumination and further functionalization of 2-alkylpyridines by yttrium catalyst. ACS Catal. 12, 13792–13804 (2022).
Zheng, H. et al. Assembly of a wheel-like Eu24Ti8 cluster under the guidance of high-resolution electrospray ionization mass spectrometry. Angew. Chem. Int Ed. 57, 10976–10979 (2018).
Liu, W. et al. Large-scale and high-resolution mass spectrometry-based proteomics profiling defines molecular subtypes of esophageal cancer for therapeutic targeting. Nat. Commun. 12, 1–18 (2021).
Pareek, V., Tian, H., Winograd, N. & Benkovic, S. J. Metabolomics and mass spectrometry imaging reveal channeled de novo purine synthesis in cells. Science 368, 283–290 (2020).
Purcell, J. M., Hendrickson, C. L., Rodgers, R. P. & Marshall, A. G. Atmospheric pressure photoionization fourier transform ion cyclotron resonance mass spectrometry for complex mixture analysis. Anal. Chem. 78, 5906–5912 (2006).
Joshi, A., Zijlstra, H. S., Collins, S. & McIndoe, J. S. Catalyst deactivation processes during 1-hexene polymerization. ACS Catal. 10, 7195–7206 (2020).
Bütikofer, A. & Chen, P. Cyclopentadienone iron complex-catalyzed hydrogenation of ketones: an operando spectrometric study using pressurized sample infusion-electrospray ionization-mass spectrometry. Organometallics 41, 2349–2364 (2022).
Oeschger, R. J., Bissig, R. & Chen, P. Model compounds for intermediates and transition states in sonogashira and negishi coupling: d 8 – d 10 bonds in large heterobimetallic complexes are weaker than computational chemistry predicts. J. Am. Chem. Soc. 144, 10330–10343 (2022).
Gubler, J., Radić, M., Stöferle, Y. & Chen, P. 2‐aminoalkylgold complexes: the putative intermediate in Au‐catalyzed hydroamination of alkenes does not protodemetalate. Chem. Eur. J. 28, e202200332 (2022).
Zhang, X. et al. Identifying metal-oxo/peroxo intermediates in catalytic water oxidation by in situ electrochemical mass spectrometry. J. Am. Chem. Soc. 144, 17748–17752 (2022).
Zhang, H. et al. Highly enantioselective construction of fully substituted stereocenters enabled by in situ phosphonium-containing organocatalysis. ACS Catal. 10, 5698–5706 (2020).
De Bruycker, K., Welle, A., Hirth, S., Blanksby, S. J. & Barner-Kowollik, C. Mass spectrometry as a tool to advance polymer science. Nat. Rev. Chem. 4, 257–268 (2020).
Baba, K. et al. Fused metalloporphyrin thin film with tunable porosity via chemical vapor deposition. ACS Appl Mater. Interfaces 12, 37732–37740 (2020).
de Jonge, N. F. et al. MS2Query: reliable and scalable MS2 mass spectra-based analogue search. Nat. Commun. 14, 1752 (2023).
Mongia, M. et al. Fast mass spectrometry search and clustering of untargeted metabolomics data. Nat. Biotechnol. 42, 1672–1677 (2024).
Zuffa, S. et al. microbeMASST: a taxonomically informed mass spectrometry search tool for microbial metabolomics data. Nat. Microbiol 9, 336–345 (2024).
Mohimani, H. et al. Dereplication of peptidic natural products through database search of mass spectra. Nat. Chem. Biol. 13, 30–37 (2017).
Kertesz-Farkas, A., Reiz, B., P. Myers, M. & Pongor, S. Database searching in mass spectrometry based proteomics. Curr. Bioinform 7, 221–230 (2012).
Haseeb, M. & Saeed, F. High performance computing framework for tera-scale database search of mass spectrometry data. Nat. Comput Sci. 1, 550–561 (2021).
Kong, A. T., Leprevost, F. V., Avtonomov, D. M., Mellacheruvu, D. & Nesvizhskii, A. I. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520 (2017).
Mao, Z., Zhang, R., Xin, L. & Li, M. Mitigating the missing-fragmentation problem in de novo peptide sequencing with a two-stage graph-based deep learning model. Nat. Mach. Intell. 5, 1250–1260 (2023).
Altenburg, T., Giese, S. H., Wang, S., Muth, T. & Renard, B. Y. Ad hoc learning of peptide fragmentation from mass spectra enables an interpretable detection of phosphorylated and cross-linked peptides. Nat. Mach. Intell. 4, 378–388 (2022).
Verheggen, K. et al. Anatomy and evolution of database search engines—a central component of mass spectrometry based proteomic workflows. Mass Spectrom. Rev. 39, 292–306 (2020).
Sun, X. et al. Omicseq: A web-based search engine for exploring omics datasets. Nucleic Acids Res. 45, W445–W452 (2017).
Gauglitz, J. M. et al. Enhancing untargeted metabolomics using metadata-based source annotation. Nat. Biotechnol. 40, 1774–1779 (2022).
Li, D. et al. XY-meta: a high-efficiency search engine for large-scale metabolome annotation with accurate FDR estimation. Anal. Chem. 92, 5701–5707 (2020).
Bach, E., Schymanski, E. L. & Rousu, J. Joint structural annotation of small molecules using liquid chromatography retention order and tandem mass spectrometry data. Nat. Mach. Intell. 4, 1224–1237 (2022).
Goldman, S. et al. Annotating metabolite mass spectra with domain-inspired chemical formula transformers. Nat. Mach. Intell. 5, 965–979 (2023).
Ludwig, M. et al. Database-independent molecular formula annotation using Gibbs sampling through ZODIAC. Nat. Mach. Intell. 2, 629–641 (2020).
Yang, Q. et al. Ultra-fast and accurate electron ionization mass spectrum matching for compound identification with million-scale in-silico library. Nat. Commun. 14, 3722 (2023).
Kuhl, C., Tautenhahn, R., Böttcher, C., Larson, T. R. & Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 84, 283–289 (2012).
Dührkop, K. et al. SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 16, 299–302 (2019).
Valkenborg, D., Mertens, I., Lemière, F., Witters, E. & Burzykowski, T. The isotopic distribution conundrum. Mass Spectrom. Rev. 31, 96–109 (2012).
Wei, Y. et al. Machine-learning-enhanced time-of-flight mass spectrometry analysis. Patterns 2, 100192 (2021).
Pluskal, T., Castillo, S., Villar-Briones, A. & Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 11, 395 (2010).
King, E., Overstreet, R., Nguyen, J. & Ciesielski, D. Augmentation of MS/MS Libraries with Spectral Interpolation for Improved Identification. J. Chem. Inf. Model 62, 3724–3733 (2022).
Degen, J., Wegscheid‐Gerlach, C., Zaliani, A. & Rarey, M. On the art of compiling and using ‘drug‐like’ chemical fragment spaces. ChemMedChem 3, 1503–1507 (2008).
van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn Res. 9, 2579–2605 (2008).
Huang, W., Bai, J., Guo, Y., Chong, Q. & Meng, F. Cobalt‐catalyzed regiodivergent and enantioselective intermolecular coupling of 1,1‐disubstituted allenes and aldehydes. Angew. Chem. Int Ed. 62, e202219257 (2023).
Li, C. et al. Cobalt‐catalyzed regio‐ and stereoselective hydroboration of allenes. Angew. Chem. 132, 6337–6342 (2020).
Guo, R. et al. Photoinduced copper‐catalyzed asymmetric C(sp3)−H alkynylation of cyclic amines by intramolecular 1,5‐hydrogen atom transfer. Angew. Chem. 134, e202208232 (2022).
Zhang, R. et al. Bio-inspired lanthanum-ortho-quinone catalysis for aerobic alcohol oxidation: semi-quinone anionic radical as redox ligand. Nat. Commun. 13, 428 (2022).
Wang, Y.-F. & Zhang, M.-T. Proton-coupled electron-transfer reduction of dioxygen: the importance of precursor complex formation between electron donor and proton donor. J. Am. Chem. Soc. 144, 12459–12468 (2022).
Lou, S.-J., Zhuo, Q., Nishiura, M., Luo, G. & Hou, Z. Enantioselective C–H alkenylation of ferrocenes with alkynes by half-sandwich scandium catalyst. J. Am. Chem. Soc. 143, 2470–2476 (2021).
Fortman, G. C. & Nolan, S. P. N-Heterocyclic carbene (NHC) ligands and palladium in homogeneous cross-coupling catalysis: a perfect union. Chem. Soc. Rev. 40, 5151 (2011).
Khazipov, O. V. et al. Fast and slow release of catalytically active species in metal/NHC systems induced by aliphatic amines. Organometallics 37, 1483–1492 (2018).
Eremin, D. B. et al. Ionic Pd/NHC catalytic system enables recoverable homogeneous catalysis: mechanistic study and application in the Mizoroki–heck reaction. Chem. – A Eur. J. 25, 16564–16572 (2019).
Eremin, D. B. et al. Mechanistic study of Pd/NHC‐catalyzed sonogashira reaction: discovery of NHC‐ethynyl coupling process. Chem. – A Eur. J. 26, 15672–15681 (2020).
Gordeev, E. G., Eremin, D. B., Chernyshev, V. M. & Ananikov, V. P. Influence of R–NHC coupling on the outcome of R–X oxidative addition to Pd/NHC complexes (R = Me, Ph, Vinyl, Ethynyl). Organometallics 37, 787–796 (2018).
Ananikov, V. P., Zalesskiy, S. S., Orlov, N. V. & Beletskaya, I. P. Nickel-catalyzed addition of benzenethiol to alkynes: formation of carbon-sulfur and carbon-carbon bonds. Russian Chem. Bull. 55, 2109–2113 (2006).
Chernyshev, V. M., Denisova, E. A., Eremin, D. B. & Ananikov, V. P. The key role of R–NHC coupling (R = C, H, heteroatom) and M–NHC bond cleavage in the evolution of M/NHC complexes and formation of catalytically active species. Chem. Sci. 11, 6957–6977 (2020).
Chernyshev, V. M. et al. Revealing the unusual role of bases in activation/deactivation of catalytic systems: O–NHC coupling in M/NHC catalysis. Chem. Sci. 9, 5564–5577 (2018).
Chagunda, I. C., Fisher, T., Schierling, M. & Mcindoe, J. S. The Poisonous Truth about the Mercury Drop Test: The Effect of Elemental Mercury on Pd(0) and Pd(II)ArX Intermediates. https://doi.org/10.26434/chemrxiv-2023-mfngl.
Frisch, M. J. et al. Gaussian 16 Revision C.01. (2016).
Ernzerhof, M. & Perdew, J. P. Generalized gradient approximation to the angle- and system-averaged exchange hole. J. Chem. Phys. 109, 3313–3320 (1998).
Petersson, G. A. & Al‐Laham, M. A. A complete basis set model chemistry. II. Open‐shell systems and the total energies of the first‐row atoms. J. Chem. Phys. 94, 6081–6090 (1991).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297 (2005).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput Chem. 32, 1456–1465 (2011).
Scalmani, G. & Frisch, M. J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 132, 114110 (2010).
Kozlov K. S. et al. Discovering organic reactions with a machine-learning-powered deciphering of tera-scale mass spectrometry data. Figshare, https://doi.org/10.6084/m9.figshare.27949029 (2025).
Kozlov K. S. et al. Discovering organic reactions with a machine-learning-powered deciphering of tera-scale mass spectrometry data. Zenodo, https://doi.org/10.5281/zenodo.14279139 (2025).
Acknowledgements
The authors thank Dr. M. Nechaev for providing Pd/NHC complexes.
Author information
Authors and Affiliations
Contributions
K.S.K. designed and implemented computational pipeline, computational experiments, and performed analysis of the results; D.A.B. designed computational pipeline, computational and experimental verification studies, and performed analysis; J.V.B. performed and supervised experimental verification studies and mass spectrometry analysis; V.V.I. assisted with mass spectrometry measurements; A.Yu.K. performed computational chemistry study; E.D.P. performed experimental verification studies; V.P.A. designed the concept, developed the idea, supervised the study and secured funding. All authors contributed to the manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Manabu Fujii and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kozlov, K.S., Boiko, D.A., Burykina, J.V. et al. Discovering organic reactions with a machine-learning-powered deciphering of tera-scale mass spectrometry data. Nat Commun 16, 2587 (2025). https://doi.org/10.1038/s41467-025-56905-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56905-8
