
Abstract
Monkeypox virus (MPXV), a zoonotic pathogen, re-emerged in 2022 with the Clade IIb variant, raising global health concerns due to its unprecedented spread in non-endemic regions. Recent studies have shown that Clade IIb (2022 MPXV) is marked by unique genomic mutations and epidemiological behaviors, suggesting variations in host-virus interactions. This study aimed to identify the differentially expressed genes (DEGs) induced by the 2022 MPXV infection through comprehensive bioinformatics analyses of microarray and RNA-Seq datasets from post-infected cell types with different MPXV clades. Subsequently, gene expression network analyses pinpoint the key DEGs, followed by their candidate drug assessment using the Drug SIGnatures DataBase (DSigDB) and validation by multiple machine learning algorithms. Comparative differential gene expression (DGE) analysis revealed 798 DEGs exclusive to the 2022 MPXV invasion in the skin cell types (keratinocytes). Intriguingly, 13 key DEGs were identified across hubs and clusters, highlighting their aberrant expressions in cell cycle regulation, immune responses, and cancer pathways. Biomarker screening via Random Forest (RF) model (selected with PyCaret from multiple models) and validation through t-distributed stochastic neighbor embedding (t-SNE) algorithm, principal component analysis (PCA), and ROC curve analysis employing Logistic Regression and Random Forest, identified 6 key DEGs (TXNRD1, CCNB1, BUB1, CDC20, BUB1B, and CCNA2) as promising biomarkers (AUC > 0.7) for clade IIb infection. This study anticipates that further investigation and clinical trials will catalyze novel detection and therapeutic options to combat 2022 MPXV infection in humans.
Similar content being viewed by others

Introduction
Following the COVID-19 pandemic, the World Health Organization (WHO) declared a Public Health Emergency of International Concern (PHEIC) in 2022 due to the re-emergence of the Mpox (previously known as monkeypox) virus (MPXV) in non-endemic regions outside of Africa including Europe, the United Kingdom, the Middle East, Australia, and North and South America1,2,3. From January 2022 to July 2024, 116 WHO Member States documented 99,176 positive cases and 208 fatalities1. Since 1 January 2022, the Democratic Republic of the Congo (DRC) reported 30,711 suspected cases and 1,326 deaths notably among children (82%)1. Around 15 countries including the USA and Spain have experienced a recent surge in MPXV infections, according to the WHO Monkeypox Dashboard. Due to this upsurge of Mpox, WHO renewed the PHEIC declaration on 14 August 20244. Given that MPXV has spread worldwide in an alarming manner, it poses a significant threat to global health and livelihoods, driving a great interest in the broader scientific community.
Unlike previous Mpox virus clades (I and IIa), a newly emerged zoonotic viral disease (clade IIb or 2022 MPXV) is classified by a complex and broader spectrum of clinical presentations, with increased infectivity5, less virulence6, and lower-case fatality rate3,6. When human keratinocytes and colon organoids were exposed to the 2022 MPXV, significant cellular damage to the skin and upregulation of hypoxia-induced genes were observed, indicating potential dysregulation of various biological functions7. Therefore, it is crucial to study the unique changes in gene expression by 2022 MPXV infection as the fundamental question of how this new clade showed a marked difference otherwise remains unanswered.
Taxonomically, the Monkeypox virus (MPXV) is a dsDNA virus classified within the Orthopoxvirus genus of the Poxviridae family, belonging to the Chordopoxvirinae subfamily. MPXV infection manifests clinically through symptoms such as headaches, myalgia, nausea, vomiting, prostration, fatigue, and malaise8. Typically, the disease manifests with an initial flu-like illness and lymphadenopathy, progressing to a widespread rash on the face and body8. With the new clade, the rash has been seen increasingly appearing in the genital regions9. Recent studies have linked MPXV infection to the disruption of NK-cell function, lymphopenia, immune evasion, cytokine storms, and impairment of the host complement system10. In severe cases, MPXV infections can result in complications including hemorrhagic manifestations, necrotic tissue damage, obstructive pathology, vital organ inflammation, and septicemia11.
To date, a group of studies on 2022 MPXV have primarily focused on the virological characterization in human- and animal-derived cell types, interactions with other diseases and viruses, and diagnostic methodologies7,12,13. Nevertheless, the complete array of computational investigations to identify exclusive differentially expressed genes (DEGs) specific to clade IIb and their roles in biological function has yet to be fully explored. This study aimed to elucidate the gene expression changes induced by the 2022 MPXV in various human cell types including monocyte, dermal fibroblast, macrophage, keratinocyte, and colon organoids. We hypothesized that certain DEGs might be responsible for alterations in biological functions and the development of critical illness and that those genes might represent biomarkers and antiviral targets specific to the 2022 MPXV infection. To test these hypotheses, we selected both microarray and RNA-Seq datasets to scrutinize the transcriptome of MPXV-infected cell types to extract exclusive DEGs. Comprehensive functional analyses were performed using various bioinformatics tools to sort the key DEGs exclusive to the 2022 MPXV infection. Additionally, identified DEGs were subjected to biomarker prediction using machine learning (ML) models and targeted for predicting the therapeutic drugs.
Methods
Data collection and normalization
The datasets of transcriptome sequencing were filtered from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) server of the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/) using the keywords: “Monkeypox”, “MPXV”, “Mpox”, “microarray”, “RNA-Seq”, “transcriptome”. Subsequently, collected datasets (GSE219036, GSE11234-GPL6762, GSE24125-GPL10912) were subjected to rigorous quality control with limma14 v3.54.2 and DESeq215 v1.38.3 in R v4.3.3 to ensure accurate comparisons of gene expression between two group of samples. Datasets associated with other organisms (e.g. Mus musculus or Macaca mulatta) were excluded from the framework of our investigation. Detailed information on the included datasets is provided in Table 1.
Identification and analysis of statistically significant DEGs
We analyzed both microarray and RNA-Seq datasets to identify highly significant DEGs. For microarray datasets, t-statistics and the limma package in R were utilized among Mock, MPXV-Zaire infections, and Killed-MPXV-Zaire infections. In the RNA-Seq dataset, a negative binomial distribution-based statistical approach, incorporating the lfcShrink16 function of the DESeq2 package in R, was employed to identify DEGs for each pairwise contrast. Genes with adjusted p-values (False Discovery Rate, FDR < 0.05) determined by the Benjamini–Hochberg false discovery rate method, and |LFC|> 1 were deemed as significant DEGs. VennDiagram17 package v1.7.3 was utilized to identify DEGs intersecting various cell types and infection conditions.
DAVID and ClueGO enrichment analysis
A robust web-based functional annotation tool, Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.8 (https://david.ncifcrf.gov/) was first utilized, and the Gene Ontology (GO) (https://geneontology.org/) outcomes were integrated with the expression data (LFC) using the GOplot v1.0.2 in R to visualize Bubble Plots for both up- and down-regulated DEGs. Significance thresholds for GO analysis were set at an adjusted p-value < 0.05 and gene count ≥ 2. Furthermore, the Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.genome.jp/kegg/) pathway analysis of the key DEGs was performed using Cytoscape18 v3.10.1 with ClueGO19 v2.5.10 and CluePedia20 v1.5.10 plugins.
Analyses of PPI networks, cluster, hub genes, and gene correlation
After grouping the DEGs, a Protein–Protein Interaction (PPI) network was constructed and analyzed using the web-based tool STRING v12.0 (https://string-db.org/). This facilitated the visualization of relationships among the up- and down-regulated genes induced by the 2022 MPXV, with a confidence interaction score threshold of ≥ 0.4 to ensure a robust PPI network. To identify highly interconnected regions within the network, the Molecular Complex Detection (MCODE)21 plugin v2.0.2 was employed, with parameters set as Kappa score (K-core) at 2, Degree Cutoff at 2, Max. Depth at 100, and Node Score Cutoff at 0.2. Hub genes were identified using the cytoHubba22 v0.1 plugin in Cytoscape. The top 10 hub genes, ranked by degree were selected, and the shortest path was displayed to highlight nodes with higher connectivity and their regulatory significance within the network. GO and KEGG terms for the hub genes were extracted using DAVID. Heatmaps were generated using TBtools-II23 v2.097, with LFC values, selecting Complete Linkage as the clustering method, the Euclidean Algorithm as the distance method, and the Cladogram as the Branch Form. Moreover, Pearson’s correlation coefficients were calculated for each gene expression pattern and visualized with a matrix diagram using the corrplot24 v0.92 package in R.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) (https://www.gsea-msigdb.org/) was conducted by comparing Clade IIb to mock samples using GSEA25 v4.3.2. The analysis utilized the built-in hallmark gene sets as the reference database and normalized RNA-Seq expression data as the input dataset. The Ensemble Gene ID platform was selected as a chip platform to map the gene expression data. The GSEA analysis identified gene sets expressed upon Clade IIb infection and extracted the DEGs that overlapped with those in the Hallmark pathway gene sets.
Probable drug prediction
To investigate pharmacological agents targeting key genes shared among cluster and hub genes of 2022 MPXV infection, the Drug Signatures database (DSigDB) (https://dsigdb.tanlab.org/) in Enrichr (https://maayanlab.cloud/Enrichr/) was utilized to find out the top 10 candidate drugs (p-value < 0.05 and combined score > 100). Outcomes were visualized with a bar plot using the ggplot226 v3.5.1 package in R. Additionally, all DEGs from each old clade (Clade IIa, Clade I, and Zaire) infection were analyzed individually to identify their candidate drugs.
Machine learning model selection, dimensionality reduction, and model evaluation
By cross-referencing the exclusive DEGs with hub genes and cluster genes, DEGs common to both analyses were selected as features for machine learning analysis. Multiple classification models were compared using the expression data of the features to categorize samples from dataset GSE219036, which includes a total of 24 samples with Clade I, Clade IIa, Clade IIb, and Mock infections.
This comprehensive analysis was conducted through PyCaret’s (https://pycaret.org/) classification module in Python v3.10.12, an automated ML library, which streamlines the end-to-end model-building process by serving as a platform for several machine learning libraries and frameworks such as XGBoost (https://xgboost.readthedocs.io/en/latest/), scikit-learn (https://scikit-learn.org/stable/), LightGBM (https://github.com/microsoft/LightGBM), and many more. We utilized PyCaret v3.3.2 along with its dependencies, including Pandas (v2.1.4), SciPy (v1.11.4), Joblib (v1.3.2), Scikit-Learn (v1.4.2), Sktime (v0.26.0), and Pmdarima (v2.0.4), among others.
Using the setup() function in PyCaret, the dataset was split into training (80%, 19 samples) and testing (20%, 5 samples) datasets. During model training, tenfold cross-validation was applied to the training data, which included default preprocessing, feature generation and feature selection. In PyCaret, the compare_models() function evaluates multiple classification algorithms to identify the best-performing model for a given dataset. The models compared include the Random Forest Classifier (RF), which is a robust ensemble learning algorithm that builds multiple decision trees during training, incorporating randomness to produce a diverse set of classifiers27. The Logistic Regression (LR) model functions as a linear model for classification tasks rather than regression in the machine learning environment28. The Extra Trees Classifier (ET) fits several randomized decision trees on various sub-samples of the dataset, enhancing robustness29. The SVM-Linear Kernel (SVM) can perform both binary and multi-class classification30, while the Ada Boost Classifier (ADA) acts as a meta-estimator that combines multiple weak classifiers to form a strong classifier31. Quadratic Discriminant Analysis (QDA) utilizes a quadratic decision boundary by modeling the class-conditional densities of the data and applying Bayes’ rule for classification32. Additionally, the K Neighbors Classifier (KN) implements the k-nearest neighbors voting mechanism33, and the Dummy Classifier (DUMMY) serves as a baseline model that makes predictions disregarding input features34. The function also evaluates more sophisticated models like Extreme Gradient Boosting (XGBoost), which employs gradient boosting algorithms, and Light Gradient Boosting Machine (LightGBM), a histogram-based framework designed for enhanced performance, alongside other available models within PyCaret.
The best-performing model, Random Forest (RF), was selected based on various performance metrics, including accuracy, AUC, recall, precision, and F1 score. We optimized the Random Forest model to enhance its performance. However, PyCaret indicated that the original model outperformed the tuned version. Consequently, the original model was retained with key hyperparameters: n_estimators = 100, criterion = ‘gini’, max_features = ‘sqrt’, bootstrap = True.
The RF model was utilized to identify the top 10 features along with their corresponding feature importance scores. Subsequently, t-SNE using Rtsne35 v0.17 package and the PCA were employed in R with that top 10 features to identify different clusters at varying scales. To evaluate the predictive ability of individual features as markers, we employed ROC analysis, calculating corresponding AUC values using LR and RF models in R, utilizing pROC36 v1.18.5 and randomForest37 v4.7.1.1 Packages.
Results
Retrieval, processing, and normalization of datasets
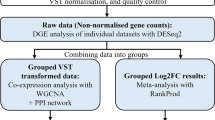
A total of three datasets, two microarrays (GSE11234-GPL6762 and GSE24125-GPL10912) and an RNA-Seq dataset (GSE219036) were retrieved from the GEO. Microarray datasets were of Zaire clade infections of two different timelines (2008 and 2011) in Dermal Fibroblast, Monocyte, and Macrophage, while the RNA-Seq dataset was of infected cell types of keratinocytes and colon organoids by MPXV clade I, IIa, and IIb. Pre-processing with quantile normalization and geometric mean scaling factor ensured cleaner, more reliable datasets, minimizing technical errors. PCA and UMAP reduced unwanted variations and enhanced the comparability of expression levels (Fig. 2, A-D). The schematic representation of our study is shown in Fig. 1.

Schematic illustration of the study.
Differential gene expression analysis highlights significant variations across cell types and MPXV clades
DGE analysis revealed a large number of significant genes (p-value < 0.05) in keratinocyte, fibroblast, and monocyte cell types, while fewer significant genes were observed in colon organoids (Fig. 2E), as illustrated in the volcano plots determined by |LogFC|> 1 and p-value < 0.05 (Fig. 3, A-F) (Supplementary Fig. 1, A & B). Within the GPL21697 platform of the GSE219036 dataset, different numbers of DEGs were found under different contrasts in both keratinocytes and colon organoids (Fig. 3, H & I). As identical DEGs were observed in two platforms, GPL10912 of GSE24125 and GPL6762 of GSE11234 datasets of different cell types such as between dermal fibroblasts and fibroblasts, as well as macrophages and monocytes, under the MPXV versus mock-infected comparison, GPL10912 of GSE24125 dataset was excluded from the downstream analyses (Fig. 3G) (Supplementary Fig. 1, C & D).

Data normalization. (A,B) PCA of RNA-Seq data highlighting clusters among the samples. X-axis and Y-axis represent the first principal component (PC1) and the second principal component (PC2), respectively. (C,D) UMAP of the microarray datasets identifying two outliers in both datasets. Each circle represents an individual, with varying colors denoting the different treatments. (E) Specific contrast depicts the distribution of adjusted p-values for each analysis, revealing a substantial number of significant genes in monocyte, fibroblast, and keratinocyte cell types.

Identification of DEGs from each contrast of the datasets. (A–F) The volcano plots illustrate the up- and down-regulated genes. The x-axis denotes the log2 fold change (LFC) in gene expression, with positive values indicating upregulation and negative values indicating downregulation. The Y-axis shows the negative log-transformed adjusted P-value. Red circles highlight upregulated genes, while blue circles indicate downregulated genes. The horizontal line marks the FDR threshold of 0.05 and the vertical lines delineate the LFC thresholds of -1 and + 1. (G–I) The number of DEGs for each contrast and the shared DEGs, offering a comprehensive overview of gene expression changes across different conditions.
Unveiling the exclusive DEGs of 2022 MPXV infection
Following comparative analysis, intersecting genes across different strains were identified via Venn diagrams (Fig. 4, A-B), displaying DEGs (Entrez IDs) from microarray datasets annotated with corresponding gene symbols. A total of 798 exclusive DEGs were successfully identified for clade IIb infection in skin cell types (both keratinocytes and fibroblast). Of these, 396 DEGs were upregulated, while 402 genes were downregulated. The LFC and Padj values of the exclusive DEGs are attached in Supplementary Table 1. A significant number of genes among the 798 exclusive DEGs does not possess any statistical significance in old strains but a few DEGs showed different expression patterns among other clades infection (Heatmaps of both Upregulated and Downregulated DEGs Showed in Supplementary Fig. 2).

Identification of exclusive DEGs to 2022 MPXV infection and their relationships among metabolic processes. (A,B) Venn diagrams illustrate the shared and unique DEGs across various clades. (C,D) The X-axis represents the Z-Score, while the -log (adj p-value) is allocated to the Y-axis. The area of the bubbles is significantly proportional to the number of DEGs (C for upregulated and D for downregulated genes) associated with the given GO terms. (E,F) Pathway enrichment analysis of exclusively expressed upregulated (E) and downregulated (F) genes. The circles represent the pathway, while the lines indicate the connection among the given pathways.
Functional enrichment analysis of 2022 MPXV-induced exclusive DEGs
The biological processes (BP) of key DEGs showed that upregulated genes are significantly enriched in transcription regulation, hypoxia response, and brain development, while downregulated genes are enriched in mitotic spindle organization, cell division, mitotic cell cycle, mitotic spindle assembly checkpoint, mitotic cytokinesis, peptide cross-linking, epidermis development, G2/M transition of the mitotic cell cycle, keratinization, and spindle assembly in female meiosis I and mitotic spindle assembly. In addition, the molecular functions (MF) of upregulated genes highlighted their roles in protein binding, small GTPase binding, identical protein binding, metal ion binding, and actin binding. In contrast, downregulated genes are predominantly enriched in protein binding. Furthermore, the cellular components (CC) indicated that exclusive DEGs are enriched in the cytosol, membrane, nucleus, cytoplasm, nucleoplasm, extracellular exosome, microtubule cytoskeleton, midbody, spindle pole, kinetochore, and spindles (Fig. 4, C-D) (Supplementary Table 2).
As for KEGG analysis, upregulated genes were significantly enriched in pathways such as Herpes simplex virus 1 infection, glycosaminoglycan biosynthesis, one-carbon pool by folate, p53 signaling, melanogenesis, adipocytokine signaling, Hippo signaling, longevity regulation, Fc-gammaR-mediated phagocytosis, glycerolipid metabolism, VEGF signaling, and colorectal cancer. On the other hand, downregulated genes were significantly enriched in the pathways consisting of prion disease, Huntington’s disease, Parkinson’s disease, cell cycle, oxidative phosphorylation, mineral absorption, proteasome, non-alcoholic fatty liver disease, diabetic cardiomyopathy, cellular senescence, oocyte meiosis, p53 signaling pathway, neurodegenerative pathways, and antigen processing and presentation (Fig. 4, E–F) (Supplementary Table 2).
Establishment of PPI network, cluster analysis, and hub gene identification
The construction of PPI networks revealed intricate complex interrelations among exclusively up- and down-regulated genes. Figure 5A presents the PPI network of 396 upregulated genes, comprising 204 nodes and 368 edges, while Fig. 5D illustrates the network of 402 downregulated genes, consisting of 291 nodes and 1849 edges. The top 3 clusters were extracted from these networks for up- and down-regulated genes (Fig. 5, B & E). Detailed information regarding these clusters, including cluster numbers, MCODE scores, node IDs, node numbers, and edge numbers, are mentioned in Fig. 5, B & E. Two hubs from each PPI network, each containing 10 hub genes mentioned in Fig. 5, C & F with their GO and KEGG terms. Subsequently, 13 DEGs were found common between the hubs and clusters (Fig. 5G). Following the identification, the correlation among the key genes of 2022 MPXV was demonstrated by a correlation plot (Fig. 5H).

Gene expression network analysis. (A & D) The assessment of the functional and physical interactions among the exclusively expressed up- and down-regulated genes. Nodes represent proteins, and edges represent the interactions among gene products. (B & E) The interconnected regions known as clusters of proteins. (A,B & D,E) The red and blue nodes indicate up- and down-regulated genes respectively, and the straight lines represent edges. (C & F) Construction of gene regulatory networks, each containing the top 10 hub genes identified from both PPI networks. (G) The heatmap delineates the expression pattern of 13 DEGs uniquely associated with infections of different clades. (H) The correlation among the 13 DEGs is shown where red and blue colors indicate positive and negative correlation respectively. LFC: Log Fold Change; NA: Not Available.
Gene set enrichment analysis highlights critical upregulation in TNF-α signaling and downregulation across multiple pathways
Gene Set Enrichment Analysis (GSEA) was conducted on 798 DEGs exclusively expressed in response to Clade IIb infection. Among 50 predefined hallmark gene sets, only 7 showed statistical significance (FDR < 0.05). Prominently, the TNF-α signaling via the NF-κB pathway exhibited the highest Enrichment Score (ES) of 0.5893 (adjusted p-value of 0.0025), indicating significant upregulation, facilitated by genes such as SLC2A3, ATP2B1, SERPINE1, VEGFA, SDC4, BHLHE40, and TGIF1. In contrast, genes including EDN1, MAP3K8, IL1A, SGK1, TNFSF9, TUBB2A, RHOB, PLK2, and SERPINB2 were downregulated, suggesting reduced activity in this signaling route. Moreover, six other pathways demonstrated negative ES scores, reflecting decreased activity or downregulation in Clade IIb relative to the mock contrast. These pathways included oxidative phosphorylation (-0.5761), E2F targets (-0.4659), IFN-α response (-0.4753), bile acid metabolism (-0.4588), KRAS signaling DN (-0.3949), and adipogenesis (-0.389). Detailed information on the overlapping exclusive DEGs and their associated hallmark pathways is provided in Table 2. Gene sets are mentioned in Supplementary Table 3.
Identification of candidate drugs
The top 10 candidates for 2022 MPXV (Clade IIb) infection were identified including Etoposide, 0173570 − 0000, Resveratrol, Methotrexate, Trifluridine, Ciclopirox, Monobenzone, Lucanthone, Cryptolepine, Trabectedin (Fig. 6A). The comparison of top 10 candidate drugs across all clade infections is presented in Table 3. Clade IIb infection has no common drugs with other clades, whereas Clade IIa and Clade I infections shared several drugs, such as Lanatoside C, Digoxin, Ouabain, Strophanthidin, Proscillaridin, Helveticoside, and Digoxigenin. In contrast, in case of the Zaire strain infection, a distinct set of drugs are found that are not shared with other clades. All drug candidate lists with associated gene sets (Drug targets) are mentioned in Supplementary Table 4.

Candidate drugs and predicted biomarkers. (A) Top 10 potential therapeutic drugs for the 13 key genes expressed in 2022 MPXV infection. The y-axis represents drug names, while the horizontal bars on the x-axis represent the combined scores, with the circles indicating the -log(P-value) for each candidate. (B) Feature importance plot for the random forest model, focusing on the top 10 most important DEGs ranked by their importance coefficient. (C,D) t-SNE and PCA based on the top 10 genes’ expressions in the RNA-Seq data. Samples associated with the keratinocyte cell line show clear distinction from that associated with colon organoids as they have not shown any differential expression. (E) The ROC curve of ranked DEGs displays AUC values for each gene predicted by both LR and RF models. t-SNE: t-distributed Stochastic Neighbor Embedding; PC: Principal Component; AUC: Area Under the ROC Curve.
Machine learning models identify potential biomarkers
Among the 13 DEGs identified in 2022 MPXV infections—namely CCNB1, CCNB2, UBE2C, KIF11, BUB1, BUB1B, CCNA2, RRM2, CDC20, AURKA, TXNRD1, CTBP2, and HSPG2—the RF model (the best-performing model evaluated by PyCaret) screened the top 10 features (DEGs), as illustrated in Fig. 6B, according to their variable importance. In t-SNE and PCA analysis, the top 10 genes of the importance plot demonstrated a marked distinction of the 2022 MPXV from other clades-infected samples and mock (Fig. 6, C & D). Intriguingly, we identified 6 DEGs common in both ML models, LR and RF with high specificity and sensitivity (AUC > 0.7) as depicted in Fig. 6E. The area under the ROC curve (AUC) values for both LR and RF are as follows: TXNRD1 (0.806, 0.838), CCNA2 (0.861, 0.787), CCNB1 (0.898, 0.727), BUB1 (0.917, 0.898), CDC20 (0.87, 0.713), and BUB1B (0.806, 0.815). These AUC values indicate that these 6 DEGs could effectively differentiate 2022 MPXV patients from other MPXV subtypes, suggesting their potential as promising biomarkers.
Discussion
The re-emergence of MPXV has spurred investigations that unveiled mutations of the new clade in virulence factors, receptor recognition proteins, and immune evasion mechanisms38. These mutations contribute to distinct epidemiological characteristics, transmission dynamics38, viral infection and replication profiles, and the severity of organ-specific diseases7. However, the identification of uniquely expressed genes by the 2022 MPXV infection in skin cell types remain unsolved. Understanding these DEGs and their altered expressions following the 2022 MPXV infection is pivotal for elucidating how the 2022 MPXV clade triggers global epidemic waves, evades immune responses, and induces distinct biological anomalies. This insight may facilitate the establishment of biomarkers and therapeutic targets to face the evolving threats of the highly mutable MPXV. In this regard, this study sought to examine three transcriptomic datasets (two microarray datasets of Zaire infection in fibroblasts, macrophages, and monocytes and an RNA-Seq dataset of Clade I, IIa, and IIb in human keratinocytes and colon organoids) to investigate the 2022 MPXV-specific exclusive DEGs. We observed significant genomic expression variations in dermal fibroblasts for Zaire infections (older clade) than that in other cell types, corroborating the previous studies that indicate the vulnerability of skin tissues to MPXVs’ invasion39,40. For the MPXV clades infections (I, IIa, and IIb), we identified a significantly higher number of DEGs in keratinocytes (4037, with 798 DEGs exclusive to clade IIb), as shown in Fig. 2H and Fig. 2C–E, compared to the DEGs identified in colon organoids, where no DEGs were observed for clade IIb (Fig. 2I). Notably, the highest number of DEGs in colon organoids was observed in clade I infections, with only 7 DEGs (Fig. 2F). The reduced number of DEGs in colon organoids (Fig. 3F) is likely due to unproductive MPXV replication, resulting in an insufficient viral load to significantly affect this cell type7. Therefore, these findings align with those of Watanabe et al., where the authors reported a tenfold increase in 2022 MPXV replication in keratinocytes compared to colon organoids, underscoring the importance of studying these predominant skin cell types7.
GO and KEGG Pathway analyses revealed significant enrichment of up- and down-regulated genes across various pathways, as illustrated in Fig. 4, thus suggesting that identified exclusive DEGs play crucial roles in the 2022 MPXV pathogenesis. Through PPI network construction, cluster analysis, hub gene identification, and GSEA, 13 key DEGs were screened from 798 exclusive DEGs based on their functional correlations. Specifically, TXNRD1, CTBP2, and HSPG2 emerged as the three common upregulated genes. Overexpression of TXNRD1 is associated with metastatic cancer development through tumor cell proliferation41 and apoptotic inhibition42. CTBP2 function as transcriptional co-repressors, binding to specific DNA sequences43 and recruiting other proteins to suppress the expression of genes such as p16INK4A and E-cadherin44, thereby facilitating epithelial-to-mesenchymal transition (EMT) and enhancing cancer cell motility and invasion45. HSPG2, linked to the negative regulation of cell death and response to hypoxia, plays a potential role in uncontrolled cell growth. These findings imply that 2022 MPXV infection might modulate cancer-associated genes, although the underlying mechanisms remain to be elucidated (Fig. 4F).
Conversely, the downregulated hub and cluster genes CCNA2, CCNB1, and CCNB2 are crucial for cell cycle regulation and the p53 signaling pathway (Fig. 4F). These genes encode essential cyclins that drive cells through various cell cycle checkpoints46. CCNA2 is pivotal for the G1/S transition, while CCNA2, CCNB1, and CCNB2 facilitate the G2/M transition46. Their downregulation indicates impaired clonal expansion, differentiation, and maturation of immune cells, leading to a weakened immune response47. Rubins et al. reported that infection of MPXV induces a stealth-like effect, effectively silencing host cellular responses to the virus. They hypothesized the involvement of complex molecular mechanisms underlying this suppression of immune activation39. In our study, as illustrated in Fig. 7, we demonstrate how MPXV-mediated downregulation of key genes disrupts immune surveillance by impairing cell cycle regulation, clonal expansion, and the maturation of immune cells. This dysregulation likely facilitates immune evasion, allowing the virus to persist undetected within the host. Other downregulated genes such as AURKA, BUB1B, and BUB1 are involved in protein serine/threonine kinase activity. (Fig. 5, C & F) BUB1 and BUB1B encode a mitotic checkpoint protein48,49 and its downregulation leads to defects in chromosome segregation during cell division and may control the mitotic checkpoint by influencing protein binding49,50. UBE2C facilitates the degradation of cell cycle-related proteins by positively regulating ubiquitin-protein ligase activity51, thereby playing a crucial role in mitotic regulation52. Similarly, the KIF11 gene, also known as the molecular motor protein, is integral to centrosome separation and the formation of bipolar mitotic spindles during mitosis53. Both genes, UBE2C and KIF11 have been demonstrated as therapeutic targets for several cancers as their downregulation control the abnormal cell growth and metastasis process/motility of the tumor cell by regulating cellular events54,55.

Association of 13 key DEGs in different pathways.
GSEA reports revealed that exclusive genes predominantly affect pathways such as TNF-α signaling via NF-κB, E2F targets, and IFN-α response. The upregulated TNF-α signaling pathway, mediated by NF-κB and crucial for immune responses, demonstrated a positive enrichment score (0.5893), suggesting that the dysregulation leads to abnormal cell proliferation as overexpression results in chronic inflammation, contributing to the regulation of communication within the tumor-promoting microenvironment56,57,58. On the other hand, a significant number of genes, including key hub genes such as CDC20, RRM2, CCNB2, BUB1B, and AURKA, were downregulated, potentially impacting the expression of E2F-related gene signatures. The hallmark E2F targets, regulated by the E2F transcription factor, play a crucial role in DNA replication, DNA damage checkpoint regulation, DNA repair mechanism, tumor suppression, apoptosis, and cell cycle progression59,60. However, the downregulation of E2F targets may disrupt the cell cycle, particularly the transition from the G1/S and S/G2 phase impeding cell division processes of immune cells47 and halting the DNA repair mechanism (through regulating the cell cycle checkpoint), apoptotic process, and tumor suppressor activities59,60. Additionally, the downregulation of IFN-α, a cytokine and signaling molecule was observed, indicating impaired immune functions and high susceptibility to viral infections. Therefore, based on these functional correlations, conducting longitudinal studies on the 2022 MPXV-infected patients at different stages will provide valuable insights into the progression of the disease and the long-term effects of the virus (Fig. 7).
Furthermore, we identified the top 10 pharmacological agents for each clade. We found some common drugs which appear for the old clades (Clade IIa, Clade I). However, Clade IIb (13 DEGs) infection has its own distinct set of potential drugs. While several drugs such as Cidofovir, Brincidofovir, Ribavirin, and KAY-2–41 were employed during the 2022 Mpox outbreak, their effectiveness has been identified with low bioavailability, resistance to MPXV strains, and adverse effects11. To ensure accurate targeting of 2022 MPXV and its infection sites, our study highlighted novel candidate drugs to refine therapeutic strategies.
ML screening uncovered the diagnostic efficacy of 13 key DEGs, providing compelling insights into the potential biomarkers for the 2022 MPXV infections. The RF model, employed via PyCaret, identified these DEGs, underscoring their importance in distinguishing the 2022 MPXV infection. Our t-SNE and PCA highlighted the discriminatory power of the top 10 RF outcomes and revealed these markers can successfully differentiate 2022 MPXV patients from other MPXV subtypes. As previous studies have demonstrated RF and LR are effective ML classification algorithms for distinguishing between various classes12,61, similarly, we employed the evaluation of RF outcomes by ROC curve analyses of LR and RF algorithms. The area under the ROC curve demonstrated high specificity and sensitivity for 6 DEGs (AUC > 0.7), out of the top 10 DEGs, suggesting the robustness and reliable means for diagnostic differentiation. Therefore, the clear distinction observed in t-SNE analysis and PCA and cross-validation by AUC values anticipates that 6 genes (TXNRD1, CCNA2, CCNB1, BUB1, CDC20, and BUB1B) could serve as pivotal biomarkers, providing a targeted approach for diagnosis and monitoring the 2022 MPXV (Clade IIb) (Fig. 7).
It should be mentioned that further experimental investigation and larger-scale datasets of human samples are necessary to corroborate our findings and elucidate the underlying mechanisms of 2022 MPXV pathogenesis, as well as to prevent and manage the deadly consequence of the mutated viral infection, like the recent catastrophic SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2).
Conclusions
This study sheds light on the distinct transcriptomic signatures of 2022 MPXV infection, highlighting 13 key DEGs and their potential roles in different biological functions including cell cycle disruption, impaired immune response, and cancer development. Furthermore, we predicted candidate drugs based on these DEGs. By applying ML algorithms, 6 DEGs have been filtered out as potential biomarkers of 2022 Mpox virus infection. In fine, our findings illuminate a new perspective that could be leveraged to combat the 2022 MPXV infection.
Data availability
The datasets analyzed in the current study are available in the GEO repository. GSE219036: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE219036; GSE11234: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE11234; GSE24125: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24125.
Code availability
The scripts are available on GitHub (https://github.com/Prokash21/2022_MPXV_Project).
Change history
09 March 2025
The original online version of this Article was revised: In the original version of this Article, the Supplementary Table 2 which was included with the initial submission, was omitted from the Supplementary Information file. The correct Supplementary Information file now accompanies the original Article.
Abbreviations
- MPXV:
-
Monkeypox virus
- DGE:
-
Differential gene expression
- DEGs:
-
Differentially expressed genes
- LFC:
-
Log2 fold change
- PCA:
-
Principal component analysis
- UMAP:
-
Uniform manifold approximation and projection
- t-SNE:
-
T-distributed stochastic neighbor embedding
- ML:
-
Machine learning
- RF:
-
Random forest
- LR:
-
Logistic regression
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the curve
References
Mpox (monkeypox) outbreak 2022 - Global. World Health Organization https://www.who.int/emergencies/situations/monkeypox-oubreak-2022 (2024). (Accessed 10 08 2024).
Kraemer, M. U. G. et al. Tracking the 2022 monkeypox outbreak with epidemiological data in real-time. Lancet Infect. Dis. 22, 941–942 (2022).
Isidro, J. et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nat. Med. 28, 1569–1572 (2022).
WHO Director-General declares mpox outbreak a public health emergency of international concern. World Health Organization https://www.who.int/news/item/14-08-2024-who-director-general-declares-mpox-outbreak-a-public-health-emergency-of-international-concern (2024). (Accessed 14 08 2024).
Bunge, E. M. et al. The changing epidemiology of human monkeypox—A potential threat? A systematic review. PLoS Negl. Trop. Dis. 16, e0010141 (2022).
Americo, J. L., Earl, P. L. & Moss, B. Virulence differences of mpox (monkeypox) virus clades I, IIa, and IIb.1 in a small animal model. Proc. Natl. Acad. Sci. U.S.A. 120, e2220415120 (2023).
Watanabe, Y. et al. Virological characterization of the 2022 outbreak-causing monkeypox virus using human keratinocytes and colon organoids. J. Med. Virol. 95, 28827 (2023).
Dou, Y. M., Yuan, H. & Tian, H. W. Monkeypox virus: past and present. World J. Pediatr. 19, 224 (2023).
Davido, B., D’Anglejan, E., Jourdan, J., Robinault, A., & Davido, G. Monkeypox 2022 outbreak: cases with exclusive genital lesions. J. Travel Med. 29, (2022).
Li, H. et al. The landscape of immune response to monkeypox virus. eBioMedicine 87, 104424 (2023).
Lu, J. et al. Mpox (formerly monkeypox): pathogenesis, prevention, and treatment. Signal Transduct. Target. Ther. 8, 458 (2023).
Cai, X., Zhou, T., Shi, W., Cai, Y. & Zhou, J. Monkeypox Virus Crosstalk with HIV: An Integrated Skin Transcriptome and Machine Learning Study. ACS Omega 8, 47283–47294 (2023).
Huo, S. et al. Development of two multiplex real-time PCR assays for simultaneous detection and differentiation of monkeypox virus IIa, IIb, and I clades and the B.1 lineage. Biosaf. Health 4, 392–398 (2022).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Zhu, A., Ibrahim, J. G. & Love, M. I. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092 (2019).
Chen, H. & Boutros, P. C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. https://doi.org/10.1186/1471-2105-12-35 (2011).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Bindea, G. et al. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009).
Bindea, G., Galon, J. & Mlecnik, B. CluePedia Cytoscape plugin: pathway insights using integrated experimental and in silico data. Bioinformatics 29, 661–663 (2013).
Hogue, C. W. & Groll, M. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 29, 137–140 (2001).
Chin, C. H. et al. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8, 1–7 (2014).
Chen, C. et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742 (2023).
Wei, T., & Simko, V. R package “corrplot”: Visualization of a Correlation Matrix (Version 0.92). (2021). Available at https://github.com/taiyun/corrplot. (Accessed 9 07 2024).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 102, 15545–15550 (2005).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2016).
Breiman, L. Random forests. Mach. Learn. 45, (2001).
Hosmer, D. W., Lemeshow, S. & Sturdivant, R. X. Applied Logistic Regression: Third Edition (2013).
Geurts, P., Ernst, D. & Wehenkel, L. Extremely randomized trees. Mach. Learn. 63, 42 (2006).
Wu, T. F., Lin, C. J., & Weng, R. C. Probability estimates for multi-class classification by pairwise coupling. J. Mach. Learn. Res. 5, (2004).
Freund, Y. & Schapire, R. E. A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 55, 199 (1997).
Franklin, J. The elements of statistical learning: Data mining, inference and prediction. Math. Intelligencer. 27, 83–85 (2005).
Goldberger, J., Hinton, G. E., Roweis, S. & Salakhutdinov, R. Neighbourhood components analysis. In Advances in Neural Information Processing Systems (ed. Goldberger, J.) (MIT Press, 2004).
Pedregosa, F. et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12, 2825 (2011).
Van Der Maaten, L. Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res. 15, 3221–3245 (2015).
Turck, N. et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 8, 12–77 (2011).
Liaw, A. & Wiener, M. Classification and regression by randomForest. R J. 2, 18–22 (2002).
Luna, N. et al. Monkeypox virus (MPXV) genomics: A mutational and phylogenomic analyses of B.1 lineages. Travel Med Infect Dis. 52, 102551 (2023).
Rubins, K. H., Hensley, L. E., Relman, D. A. & Brown, P. O. Stunned silence: Gene expression programs in human cells infected with monkeypox or vaccinia virus. PLoS One 6, e15615 (2011).
Rubins, K. H., et al. Comparative analysis of viral gene expression programs during poxvirus infection: A transcriptional map of the vaccinia and monkeypox genomes. PLoS One. 3, (2008).
Yoo, M. H., Xu, X. M., Carlson, B. A., Gladyshev, V. N., & Hatfield, D. L. Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J. Biol. Chem. 281, (2006).
Tang, B., Zhang, Q., Liu, K., & Huang, Y. Exosomal circRNA FNDC3B promotes the progression of esophageal squamous cell carcinoma by sponging miR-490–5p and regulating thioredoxin reductase 1 expression. Bioengineered. 13, (2022).
Thio, S. S. C., Bonventre, J. V. & Hsu, S. I. H. The CtBP2 co-repressor is regulated by NADH-dependent dimerization and possesses a novel N-terminal repression domain. Nucleic Acids Res. 32, 1836–1847 (2004).
Yang, X. et al. C-terminal binding protein-2 promotes cell proliferation and migration in breast cancer via suppression of p16INK4A. Oncotarget. 8, 26154–26168 (2017).
Chinnadurai, G. The transcriptional corepressor CtBP: A foe of multiple tumor suppressors. Cancer Res. 69, 731–734 (2009).
Sowinska, W. et al. Regnase-2 inhibits glioblastoma cell proliferation. Sci Rep. 14, 1574 (2024).
Laphanuwat, P. & Jirawatnotai, S. Immunomodulatory Roles of Cell Cycle Regulators. Front. Cell Dev. Biol. 7, 23 (2019).
Williams, G. L., Roberts, T. M. & Gjoerup, O. V. Bub1: escapades in a cellular world. Cell Cycle 6, 1699–1704 (2007).
Tang, X. et al. BUB1B and circBUB1B_544aa aggravate multiple myeloma malignancy through evoking chromosomal instability. Sig. Transduct. Target. Ther. 6, 361 (2021).
Myrie, K. A., Percy, M. J., Azim, J. N., Neeley, C. K. & Petty, E. M. Mutation and expression analysis of human BUB1 and BUB1B in aneuploid breast cancer cell lines. Cancer Lett. 152, 193–199 (2000).
Hao, Z., Zhang, H. & Cowell, J. Ubiquitin-conjugating enzyme UBE2C: molecular biology, role in tumorigenesis, and potential as a biomarker. Tumour Biol. 33, 723–730 (2012).
Chen, Z. & Wang, L. The clinical significance of UBE2C gene in the progression of renal cell carcinoma. Eur. J. Histochem. 65, 2 (2021).
Yu, Y. & Feng, Y. M. The role of kinesin family proteins in tumorigenesis and progression: potential biomarkers and molecular targets for cancer therapy. Cancer 116, 5150–5160 (2010).
Xiong, Y. et al. UBE2C functions as a potential oncogene by enhancing cell proliferation, migration, invasion, and drug resistance in hepatocellular carcinoma cells. Biosci. Rep. 39, 4 (2019).
Zhou, J. et al. KIF11 functions as an oncogene and is associated with poor outcomes from breast cancer. Cancer Res. Treat. 51, 1207–1221 (2019).
Cabal-Hierro, L. & Lazo, P. S. Signal transduction by tumor necrosis factor receptors. Cell. Signal. 24, 1297–1305 (2012).
Balkwill, F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 25, 409–416 (2006).
Buhrmann, C. et al. Evidence that TNF-β induces proliferation in colorectal cancer cells and resveratrol can down-modulate it. Exp. Biol. Med. 244, 1–12 (2019).
Kurayoshi, K. et al. The key role of E2F in tumor suppression through specific regulation of tumor suppressor genes in response to oncogenic changes. Gene Express. Regulat. Mammalian Cells-Transcription Toward Establish. Novel Therapeut. https://doi.org/10.5772/intechopen.72125 (2018).
Bracken, A. P., Ciro, M., Cocito, A. & Helin, K. E2F target genes: unraveling the biology. Trends Biochem. Sci. 29, 409–417 (2004).
Gao, X. et al. Genome-wide screening of SARS-CoV-2 infection-related genes based on the blood leukocytes sequencing data set of patients with COVID-19. J. Med. Virol. 93, 5544–5554 (2021).
Acknowledgements
In the struggle of understanding the language of life, we acknowledge the debt of July 2024 when we all spoke the same language taught by sacrifice. We also deliver our gratitude to BMB department of SUST.
Author information
Authors and Affiliations
Contributions
Software: JP Debnath, K Hossen; Methodology: JP Debnath, K Hossen, SB Sayed, MS Khandaker, PC Dev, S Sarker, T Hossain; Data Curation: JP Debnath, K Hossen, SB Sayed, MS Khandaker, PC Dev, S Sarker; Formal analysis: JP Debnath, K Hossen, SB Sayed, MS Khandaker; Investigation: JP Debnath, K Hossen, SB Sayed, MS Khandaker; Validation: JP Debnath, K Hossen, SB Sayed, MS Khandaker, PC Dev; Visualization: JP Debnath, K Hossen, SB Sayed, MS Khandaker; Writing – original draft: JP Debnath, K Hossen, SB Sayed, MS Khandaker; Conceptualization: PC Dev, S Sarker, T Hossain; Resources: PC Dev, S Sarker, T Hossain; Supervision: PC Dev, S Sarker, T Hossain; Writing – review & editing: PC Dev, S Sarker, T Hossain; Project administration: T Hossain. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Debnath, J.P., Hossen, K., Sayed, S.B. et al. Identification of potential biomarkers for 2022 Mpox virus infection: a transcriptomic network analysis and machine learning approach. Sci Rep 15, 2922 (2025). https://doi.org/10.1038/s41598-024-80519-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-80519-7
